
lnhibition of KU70 and KU80 by CRlSPR interference, not NgAgo interference, increases the ef ficiency of homologous recombination in pig fetal fibroblasts
2019-02-14 03:12:38LlGuolingQUANRongWANGHaoqiangRUANXiaofangMOJianxinZHONGCuiliYANGHuaqiangLlZicongGUTingLlUDewuWUZhenfangCAlGengyuanZHANGXianwei
Ll Guo-ling, QUAN Rong, WANG Hao-qiang, RUAN Xiao-fang, MO Jian-xin, ZHONG Cui-li, YANG Huaqiang, , Ll Zi-cong, GU Ting, LlU De-wu, WU Zhen-fang, , CAl Geng-yuan, , ZHANG Xian-wei,
1 National Engineering Research Center for Breeding Swine Industry, College of Animal Science, South China Agricultural University, Guangzhou 510642, P.R.China
2 Wens Foodstuff Group Co., Ltd., Yunfu 527400, P.R.China
Abstract Non-homologous end-joining (NHEJ) is a predominant pathway for the repair of DNA double-strand breaks (DSB). It inhibits the ef ficiency of homologous recombination (HR) by competing for DSB targets. To improve the ef ficiency of HR, multiple CRISPR interference (CRISPRi) and Natronobacterium gregoryi Argonaute (NgAgo) interference (NgAgoi) systems have been designed for the knockdown of NHEJ key molecules, KU70, KU80, polynucleotide kinase/phosphatase (PNKP),DNA ligase IV (LIG4), and NHEJ1. Suppression of KU70 and KU80 by CRISPRi dramatically promoted (P<0.05) the ef ficiency of HR to 1.85- and 1.58-fold, respectively, whereas knockdown of PNKP, LIG4, and NHEJ1 repair factors did not significantly increase (P>0.05) HR ef ficiency. Interestingly, although the NgAgoi system significantly suppressed (P<0.05)KU70, KU80, PNKP, LIG4, and NHEJ1 expression, it did not improve (P>0.05) HR ef ficiency in primary fetal fibroblasts.Our result showed that both NgAgo and catalytically inactive Cas9 (dCas9) could interfere with the expression of target genes, but the downstream factors appear to be more active following CRISPR-mediated interference than that of NgAgo.
Keywords: homologous recombination, non-homologous end-joining, CRISPRi, NgAgoi, KU70, KU80
1. lntroduction
Gene editing by homologous recombination (HR) is a powerful tool for precise genetic modification, but HR is less active than non-homologous end joining (NHEJ) in mammalian cells, and it only occurs during the S and G2 phases, whereas NHEJ occurs throughout the entire cell cycles (Linet al. 2014). NHEJ is a major pathway for double-strand break (DSB) repair, which competitively inhibits HR ef ficiency in mammalian cells. NHEJ includes alternative NHEJ and classical NHEJ pathways (Changet al. 2017). The classical NHEJ repair pathway is initiated by the recognition and binding of the KU70/80 heterodimer to the DSB, leading to recruitment of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) (Alshareedaet al. 2013; Brittonet al. 2013), which phosphorylates and binds to the Artemis protein. Afterward, the Artemis/DNA-PKcs complex removes 5´ and 3´ DNA overhangs to create blunt ends (Gottlieb and Jackson 1993; Maet al.2002). In this process, polynucleotide kinase/phosphatase(PNKP) plays an important role in the repair of DSBs(Jilaniet al. 1999). Once the blunt ends are created, the XRCC4/DNA ligase IV/XRCC4-like factor (XLF or NHEJ1)ligation complex is recruited to join the DNA ends together(Wuet al. 2009; Hammelet al. 2011). In previous studies,several strategies have been reported, including NHEJ inhibition through either RNA interference (Chuet al.2015; Liet al. 2018) or with small molecules (Maruyamaet al. 2015), to improve HR ef ficiency in mammalian cell lines. Chuet al. (2015) and Liet al. (2018) showed that suppression of the key NHEJ pathway protein KU70 and DNA ligase IV by short hairpin RNA or siRNA can ef ficiently improve homology-directed repair ef ficiency in human,mice, and pig cell lines. Bertoliniet al. (2009) also showed that knockdown ofKU70andXRCC4in HCT116 cells significantly reduced the ef ficiency of random integration and improved the ef ficiency of site-specific integration by about 3–4 folds.
Recent studies have shown that catalytically inactive Cas9 (dCas9), which is deficient in nucleolytic activityin vitro, can ef ficiently silence target genes (Rosenbluhet al. 2017). This CRISPR interfering system, called CRISPRi, works as an orthogonal system in diverse organisms, including bacterial and human cells. In addition, a recent report of genome editing usingNatronobacterium gregoryiArgonaute (NgAgo) with guide DNA (gDNA) in zebra fish showed the ef ficient knockdown offabp11a(Qiet al. 2016); this knockdown system is referred to as NgAgoi. Compared with RNAi technology, CRISPRi and NgAgoi systems have the higher ef ficiency in gene silencing, and they render less toxicity to the target cells or organisms for low off-target effect.CRISPRi and NgAgoi systems have broad applications in various biological research areas including identifying disease-related targets, modulating gene transcription,and developing transgenic animals.
Improving HR ef ficiency in pig primary cells can facilitate the ef ficient creation of genetically engineered pigs with precise gene modifications.NHEJ1andPNKPare also crucial in NHEJ repair. It is yet uncertain whether knockdown of NHEJ1 and PNKP can promote HR ef ficiency in pig cells. We hypothesized that HR ef ficiency could be improved through inhibiting the expression of the NHEJ key factorsKU70,KU80,PNKP,NHEJ1, andLIG4.Therefore, in the present study, we compared CRISPRi system with NgAgoi system targeting multiple NHEJ factors to suppress their expression, and investigated the effects of gene silencing on HR frequency in pig primary fetal fibroblasts in order to test the above hypothesis.
2. Materials and methods
2.1. Construction of CRlSPRi system, NgAgoi system, and HDR-GFP reporter
The CRISPRi system contained gRNA vectors and dCas9 plasmid. To construct gRNA vectors for CRISPRi system,the single gRNAs (sgRNAs) targeting core regulatory region of the key NHEJ pathway factors, likeKU70,KU80,LIG4,NHEJ1, orPNKPwere designed using CRISPR-ERA(http://crispr-era.stanford.edu/index.jsp) (Appendix A and Fig. 1-A). For each gene, we designed three candidate sgRNAs according to the targeting site. The sgRNAs were synthesized as DNA oligonucleotides and cloned into the gRNA cloning vector to form CRISPRi system. The PBCAG-SKOM (dCas9) plasmid was synthesized by GenScript Co. (Nanjing, China) (Fig. 1-B).
The NgAgoi system consisted of gDNA and NLSNgAgo plasmid. The NLS-NgAgo plasmid (Fig. 1-B) was synthesized by GenScript Co. (Nanjing, China) (Fig. 1-B)and the gDNA targeting exons ofKU70,KU80,LIG4,NHEJ1, orPNKPwere designed according to protocols of Qiet al. (2016). Three candidate gDNAs with 5´phosphorylation (5´p) per gene were synthesized by the Beijing Genomics Institute (Beijing, China) (Appendix B and Fig. 1-C).
HDR-GFP reporter system contained a GFP-deficient vector and a pMD18T-GFP vector. The former was constructed by dividing the GFP-coding sequence into two segments separated by a stop codon, aBamHI and aHindIII restriction sites. The latter was constructed by inserting a promoter-free GFP gene into pMD18-T plasmid(TaKaRa, Dalian, China). The pMD18T-GFP cannot express GFPper sedue to the lack of promoter. When the two plasmids recombine to restore GFP function, the cells regain fluorescence expression (Fig. 1-D).
2.2. Experimental design and treatments
A completely randomized design was adopted in three experiments. In Exp.1, for detecting the silencing effect of CRISPRi system or NgAgoi system on each gene, 12 wells were randomly divided into four groups. Each group with three repeats was co-transfected with dCas9 plasmids and gRNA, or NgAgo plasmid and gDNA corresponding to the target site of each gene (Tables 1 and 2). In Exp.2,for assessment of HR ef ficiency induced by CRISPRi system or NgAgoi system, 18 wells were randomly divided into six groups. Each group with three repeats was cotransfected with dCas9 plasmids, optimal gRNA and HDR-GFP reporter plasmid, or NgAgo plasmid, optimal gDNA and HDR-GFP reporter plasmid (Tables 3 and 4).In Exp.3, for assessment of the changes of key factors in the NHEJ and HR pathways, nine wells were randomly divided into three groups on each system. Each group with three repeats was co-transfected with dCas9 plasmids and optimal gRNA, or NgAgo plasmid and optimal gDNA,targetingKU70orKu80gene (Table 5).
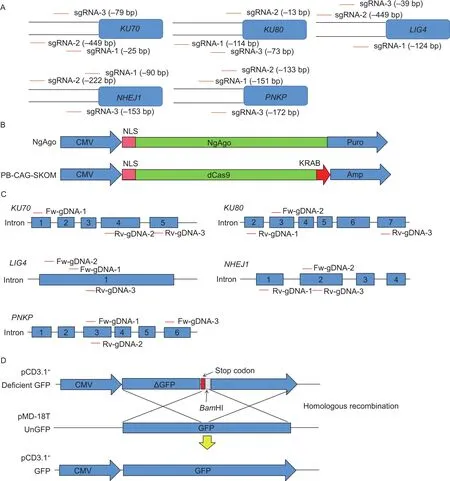
Fig. 1 CRISPR interference (CRISPRi) and Natronobacterium gregoryi Argonaute (NgAgo) interference (NgAgoi) systems,and HDR-GFP reporter. A, the specific sgRNAs were designed to target the upstream regulatory region of KU70, KU80, PNKP,LIG4, and NHEJ1 using CRISPR-ERA. B, construction of the PB-CAG-SKOM (dCas9) and NLS-NgAgo plasmids. C, guide DNAs (gDNAs) were designed to target exons of KU70, KU80, PNKP, LIG4, and NHEJ1. D, construction of the HDR-GFP reporter system, which comprised a GFP-deficient vector and pMD18T-GFP vector.
2.3. Cell culture and transfection (Exps.1, 2 and 3)
Porcine fetal fibroblasts (PFFs) were isolated from 32-dayold fetuses, which were cultured in Dulbecco’s Modi fied Eagle Medium (DMEM) (Thermo Fisher Scientific, Waltham,MA, USA) supplemented with 10% fetal bovine serum(Thermo Fisher Scientific, Waltham, MA, USA).
For examination of gene silencing effect, the PFFs were electroporated by Amaxa Nucleofector (Lonza, Basel,Switzerland) program A-033 according to the manufacturer’s instructions (Lonza, Basel, Switzerland). Brie fly, (0.5–1)×106cells were harvested, centrifuged and then resuspended in 100 μL nucleofector solution, which contains 4 μg pEGFP-N1 for transfection ef ficiency, or 8 μg dCas9 plasmid and 4 μg gRNA cloning vector, or 8 μg NgAgo vector and 3 μL 100 μmol L–15´p-gDNA. Afterward, the cell/plasmid mixture was transferred into a cuvette for nucleofection and further cultured in 6-well plates for subsequent assays (Tables 1 and 2).
For assays of HR ef ficiencies, after electrotransfection with CRISPRi or NgAgoi for 48 h, the PFFs were transfected with 2.5 μg/well HDR-GFP reporter (1.25 μg linearized GFP-deficient reporter plasmid and 1.25 μg pMD18T-GFP plasmid as HR-repair template) by Lipofectamine™ LTX Reagent with PLUS Reagent (Thermo Fisher Scientific,Waltham, MA, USA). Linearized pEGFP-N1 was used as the positive control (Tables 3 and 4). When cells were cultured for 48 h, GFP expression was monitored using an Accuri C6 Flow Cytometer (BD Biosciences, San Jose, CA,USA). The percentages of GFP expression cells represent the HR ef ficiencies.
2.4. Detection the gene silencing effect of CRlSPRi system or NgAgoi system (Exp.1)
Total RNA was isolated from PFF cells using the RNeasyMicro Kit (Qiagen, Hilden, Germany) at 48 h after electroporation. The cDNA was synthesized using the PrimeScript RT Reagent Kit (TaKaRa, Dalian, China)following the manufacturers’ protocols. The primer sequences forKU80,KU70,LIG4, NHEJ1PNKP,β-actin,are shown in Appendix C. The real-time PCR were performed in 10 μL reaction solution containing 5 ng cDNA,0.25 μmol L–1of each forward and reverse primer, and 5 μL SYBR Green I Master (Thermo Fisher Scientific, Waltham,MA, USA) using the EcoTMReal-Time PCR System (Illumina,San Diego, CA, USA). The thermal cycle was 95°C for 5 min; followed by 45 cycles at 95°C for 10 s, 60°C for 10 s,and 72°C for 10 s. The expression levels of tested genes were calculated by normalizing to β-actin level.
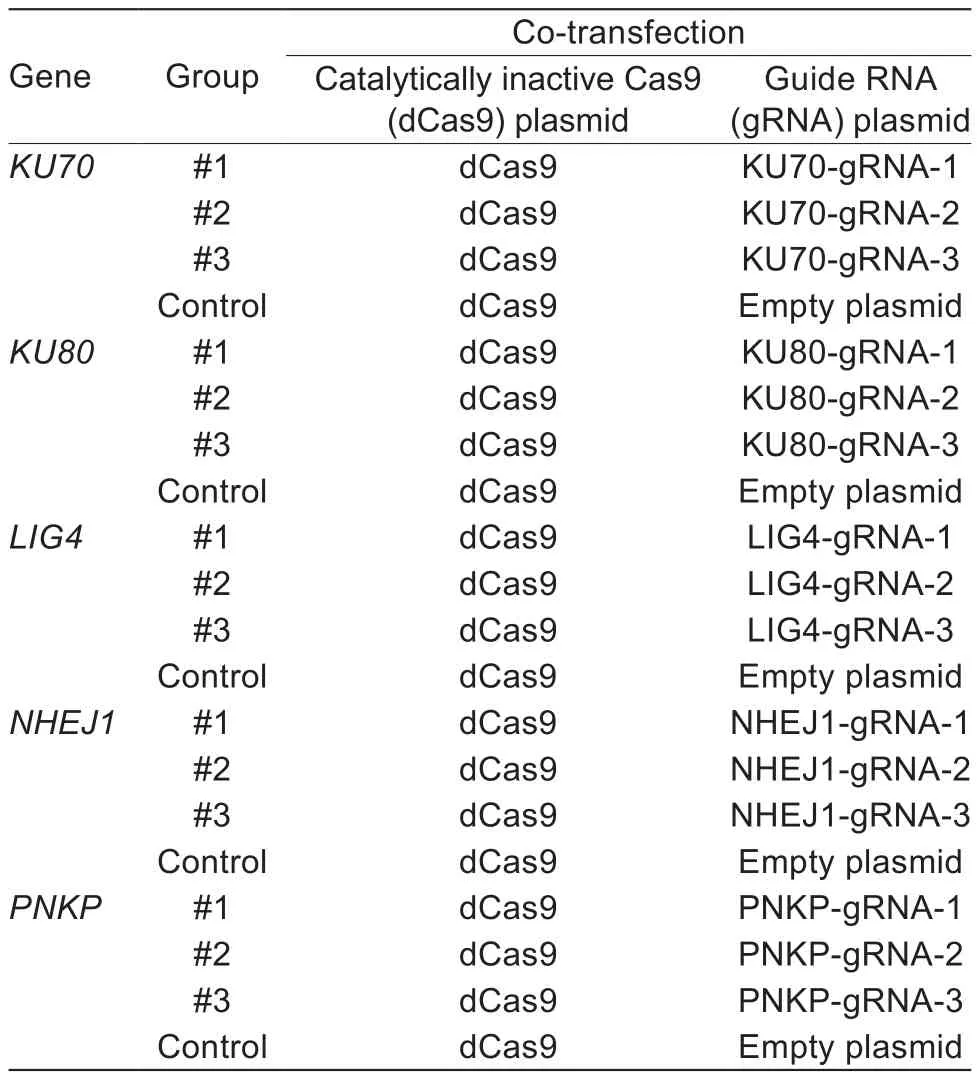
Table 1 Treatment with CRISPR interference (CRISPRi)system (Exp.1)
2.5. T7 endonuclease l assay (Exp.2)
The target sites of the best-functioning CRISPRi and NgAgoi systems were PCR-ampli fied using the primer sets listed in Appendix D. The PCR reaction was performed to amplify the target sequences from a 200 ng of genomic DNA templates with a PrimeSTAR Max DNA Polymerase(TaKaRa, Dalian, China) according to the manufacturer’s protocol. The thermocycler setting consisted of one cycle at 98°C for 5 min; 30 cycles at 98°C for 10 s, 62°C for 5 s,and 72°C for 10 s; and one cycle of 72°C for 5 min. Thenthe PCR products were denatured and reannealed as the following thermocycler settings: 95°C, 10 min; 95–85°C at–2°C s–1; 85°C for 1 min; 85–75°C at –0.1°C s–1; 75°C for 1 min; 75–65°C at –0.1°C s–1; 65°C for 1 min; 65–55°C at–0.1°C s–1; 55°C for 1 min; 55–45°C at –0.1°C s–1; 45°C for 1 min; 45–35°C at –0.1°C s–1; 35°C for 1 min; 35–25°C at –0.1°C s–1; 25°C for 1 min; and hold at 4°C. Afterward,5 U of T7 endonuclease I (NEB, Ipswich, MA, USA) was added to digest the re-annealed DNA at 37°C for 30 min.The reaction products were resolved on a 3% agarose gel.The relative intensities of DNA bands were quantitated using Image Lab (Bio-Rad, Hercules, CA, USA). Cleavage activity was calculated as: {1–[1–(b+c/a+b+c)]1/2}×100, where, “a”is the band intensity of the DNA substrate, and “b” and “c”are the cut products.

Table 2 Treatment with Natronobacterium gregoryi Argonaute(NgAgo) interference (NgAgoi) system (Exp.1)
2.6. Assessment of HR ef ficiency (Exp.2)
HR ef ficiency was determined by flow cytometry after transfecting PFF cell lines with HDR-GFP reporter and CRISPRi or NgAgoi system in two completely randomized experiments. PFF cell lines were randomly divided into six groups in six-well plates. Each group, with three repeats(1×106cells/repeat), was randomly assigned to one of six treatments (Tables 3 and 4).
2.7. Western blot (Exp.3)
Cell samples were homogenized in RIPA buffer, and then the supernatants were collected by centrifugation at 10 000×g for 5 min. The protein concentration was measured by BCA Protein Assay Kit (Thermo Fisher Scientific). For Western blot, 30 μg of whole cell lysate was used for SDS-PAGE and then transferred to a PVDF membrane. Afterward,the membranes were blocked with 5% non-fat dry milk in TBST for 3 h at room temperature and incubated overnight at 4°C with KU70 (MBS275894, Mybiosource) or KU80 (LSC31766, Lifespan) rabbit polyclonal antibodies. After the membranes were washed with TBST, the secondary HRP-conjugated goat anti-rabbit IgG antibody was incubated for 2 h at room temperature and imaged using a SuperSignal West Pico Enhanced Chemiluminescence Kit (Thermo Fisher Scientific). The β-actin antibody (60008-1-Ig;Proteintech, USA) was used as a loading control.
2.8. Detection of expression changes of the NHEJ and HR pathways factors (Exp.3)
PFFs cell were electroporated with CRISPRi (KU70-gRNA-3 and KU80-gRNA-2, respectively) or NgAgoi(KU70-5´p-gDNA-2 and KU80-5´p-gDNA-2, respectively).Each treatment was performed using three biological replicates with three technical replicates. The expression level ofRad50,Rad51,RPA3,PNKP,XRCC4, andLIG4were assayed using real-time PCR methods as previously described in Section 2.4. The primer sequences are shown in Appendix C. Expression levels of target genes were normalized to the housekeeping geneβ-actin.
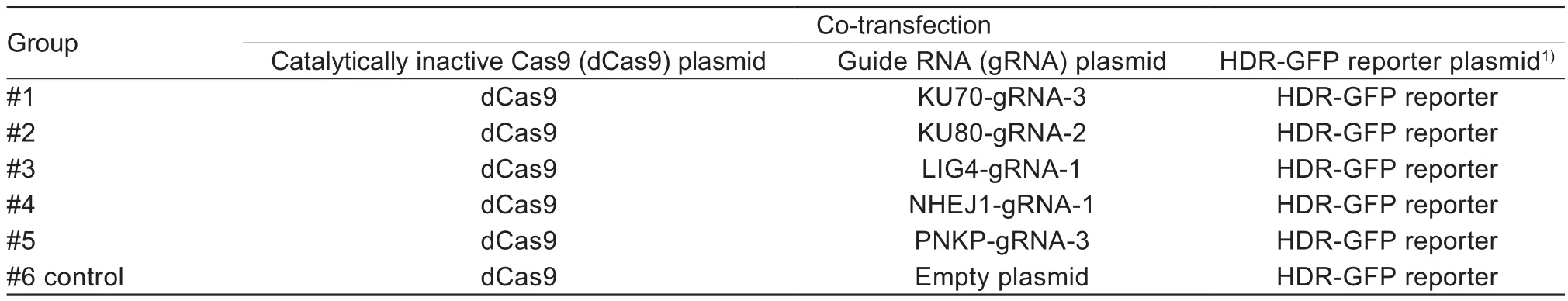
Table 3 Assessment of homologous recombination (HR) ef ficiency of cells treated with CRISPR interference (CRISPRi) system(Exp.2)
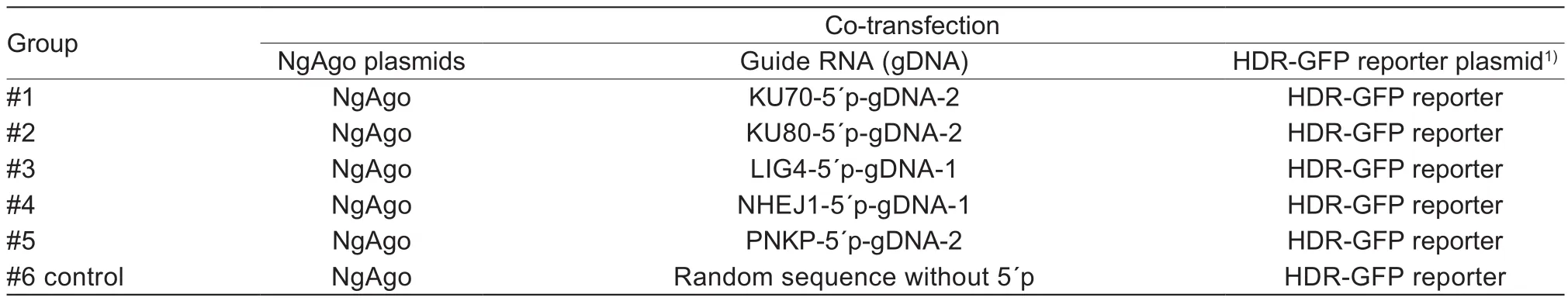
Table 4 Assessment of homologous recombination (HR) ef ficiency of cells treated with Natronobacterium gregoryi Argonaute(NgAgo) interference (NgAgoi) system (Exp.2)
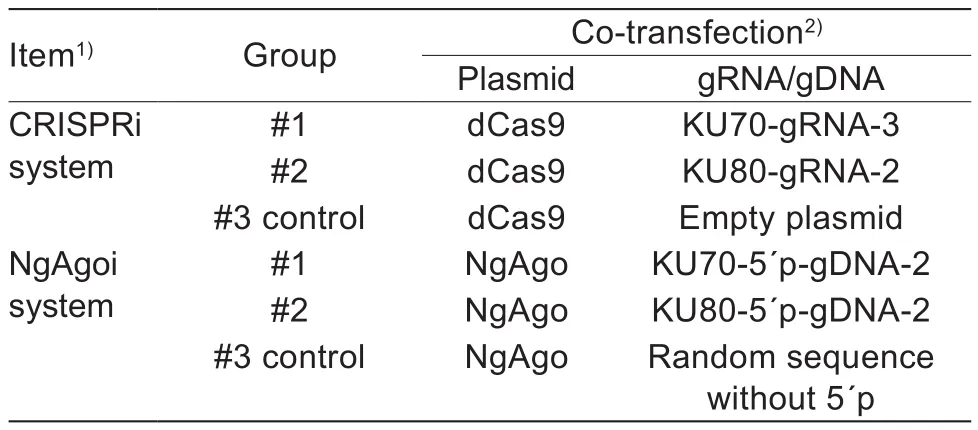
Table 5 Expression changes of key factors of cells treated with CRISPRi and NgAgoi systems (Exp.3)
2.9. Statistical analysis
The data were analyzed using the PASW Statistics 21(IBM SPSS, Chicago, IL, USA) or GLM procedure (SAS Inst. Inc., Cary, NC, USA). The replicate well served as an experimental unit. For gene expression and HR ef ficiency,one-way ANOVA analysis was used to determine the effects of treatments on results, and the differences among treatment means were analyzed by applying the Duncan’s multiple pange test. For data with unequal variances,Welch’s ANOVA followed by Games-Howell test was performed by SPSS. For data that excludes outliers, SAS GLM procedure was employed to analyze the differences among group means. All analyzed results are presented as mean±SE. The value ofP<0.05 was considered significant.
3. Results
3.1. CRlSPRi and NgAgoi knockdown of NHEJ key molecules (Exp.1)
The mRNA levels of NHEJ and HR key factors were detected in porcine fetal fibroblasts at 48 h after electroporation with CRISPRi or NgAgoi. CRISPRi (Fig. 2-A) and NgAgoi(Fig. 2-B) were able to significantly suppress (P<0.05) the corresponding gene expression with sgRNA or 5´p-ssDNA.Although almost all of the 5´p-ssDNA in the NgAgoi system significantly suppressed (P<0.05) gene expression as indicated by comparison to control cells. There were no visible differences (P>0.05) between the positive and negative strands. In addition, the CRISPRi system did not seem ideal compared with NgAgoi, particularly with regard toKU70,KU80, andPNKP, as it only knocked down about 50% of gene expression and only about two-thirds of the designed sgRNAs were effective. To con firm that the effect of suppression of CRISPRi and NgAgoi is not caused by genetic mutations, we assess their cleavage activity.
We sequenced the PCR products and the sequencing results showed that dCas9 and NgAgo cannot cleave the endogenous target locus (Fig. 2-C).
3.2. Knockdown of NHEJ key factors increased HR ef ficiency (Exp.2)
Flow cytometry assays showed that electroporation with optimal functioningKU70-sgRNA-3,KU80-sgRNA-2,LIG4-sgRNA-1, andNHEJ1-sgRNA-1PNKP-sgRNA-3 CRISPRi system, and only knockdown ofKU70andKU80significantly improved (P<0.05) HR ef ficiency to 185 and 158%, respectively (Fig. 3-A and B), whereas knockdown ofPNKP,LIG4, andNHEJ1repair factor did not increase(P>0.05) HR ef ficiency (Fig. 3-C and D). Although in the NgAgoi system, gDNAs ofKU70Rv-gDNA-2,KU80Rv-gDNA-2,LIG4Fw-gDNA-1,NHEJ1Rv-gDNA-1, andPNKPRv-gDNA-2 showed significantly knocked down(P<0.05) gene expression, it was ineffective (P>0.05)for increasing HR ef ficiency. Since the results produced under the two methods differ, we further assessed KU70 and KU80 protein expression levels by Western blot.However, the result showed that KU70 and KU80 protein demonstrated significant down-regulation under target NgAgoi or CRISPRi treatment in comparison with the control (Fig. 3-E).
3.3. Effects of interference on key genes expression in the NHEJ and HR pathways (Exp.3)
We measured the mRNA levels ofRad50,Rad51,RPA3,PNKP,XRCC4, andLIG4after electroporation with CRISPRi and NgAgoi in theKU70andKU80regions. After interference, the downstream gene expression ofKU70andKU80showed the same pattern. In the CRISPRi system,LIG4andPNKPshowed more expression (P<0.05) than those in control cells, but there was less (P<0.05)XRCC4expression than that in control cells (Fig. 4). The key factors of HR, such asRad50andRPA3, demonstrated significant upregulation (P<0.05), and onlyRad51was not affected(P>0.05). However, in the NgAgoi system, interference withKU70andKU80appeared to significantly decrease (P<0.05)the expression ofLIG4andPNKP, whileRad50,Rad51,RPA3, andXRCC4were not affected (P>0.05) (Fig. 4).
4. Discussion
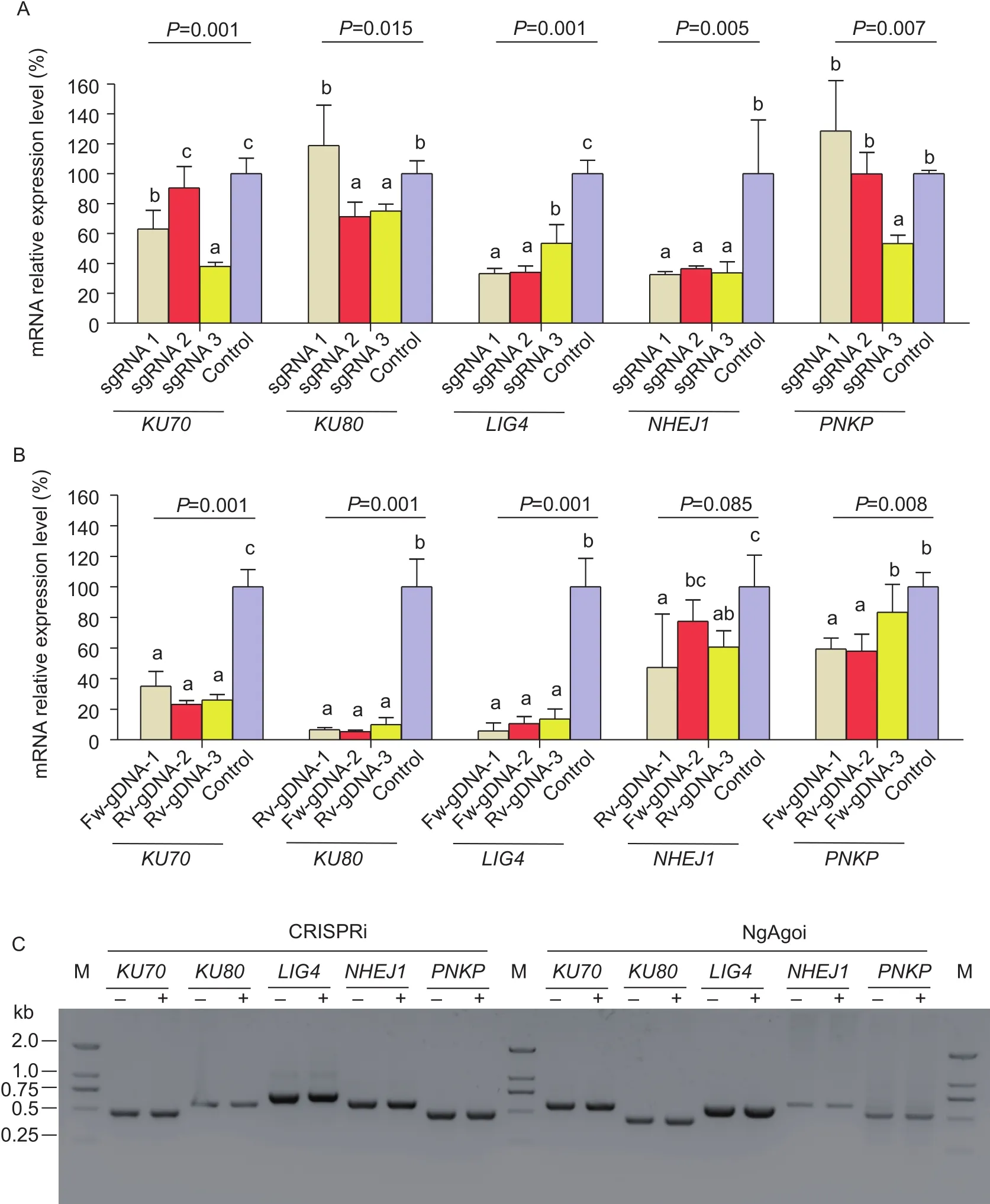
Fig. 2 Expression of non-homologous end-joining (NHEJ) factors and cleavage activity after CRISPR interference (CRISPRi)and Natronobacterium gregoryi Argonaute (NgAgo) interference (NgAgoi) systems (Exp.1). Effects of the key factors genes in the NHEJ pathway was downregulated by CRISPRi (A) or NgAgoi system (B). Fw is here represented as positive strand and Rv as a negative strand. The control was co-electroporation of sgRNA empty vector and dCas9 plasmid or electroporation of NgAgo plasmid and ssDNA. Data are mean±SE (n=3). Different letters have significant difference at P<0.05 level. C,assessment of dCas9 and NgAgo cleavage activity by T7E1 assays. + means gRNA/GDNA corresponding to the target site of each gene was contained in CRISPRi/NgAgoi systems, and – means control group (see Tables 1 and 2).
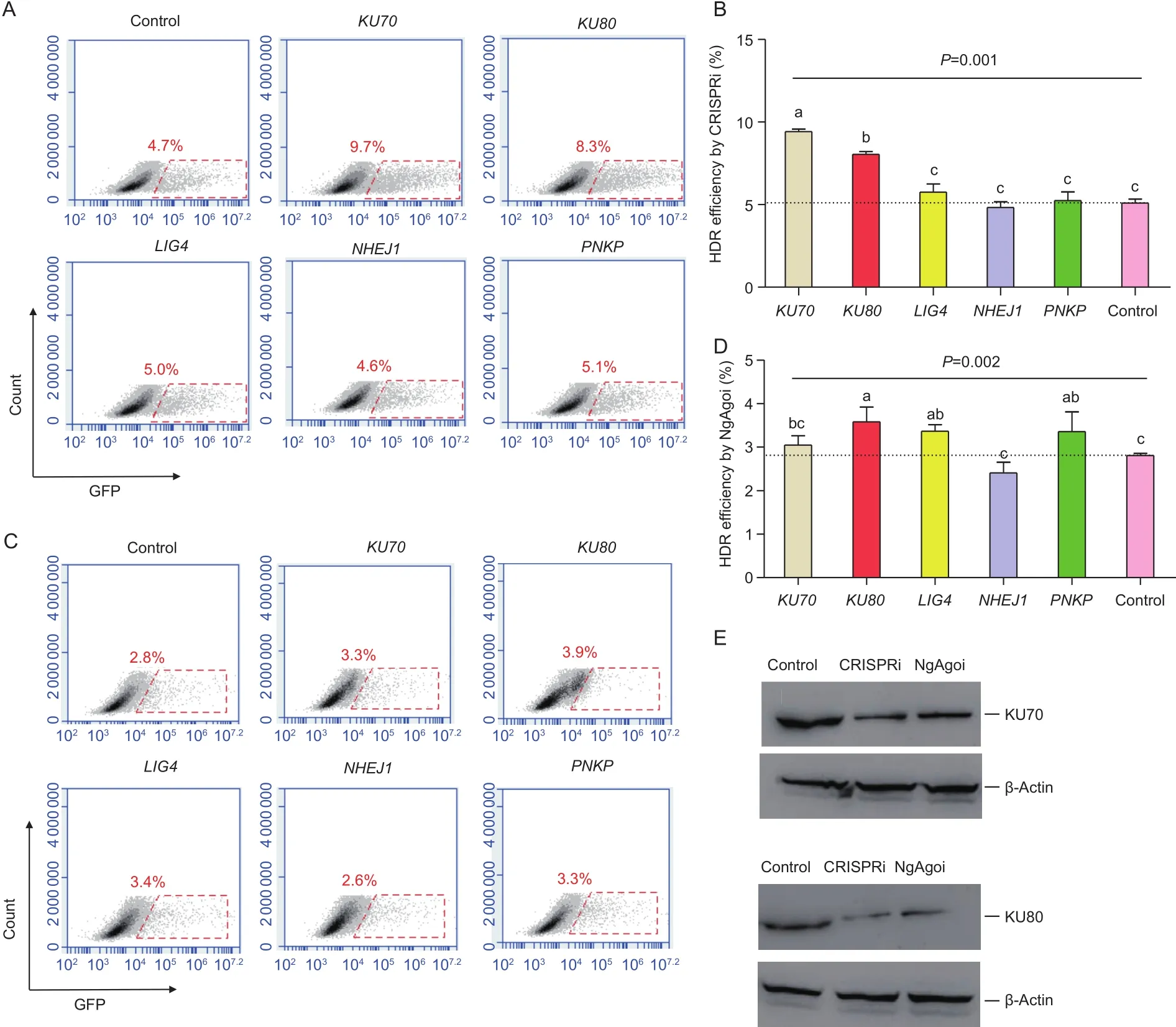
Fig. 3 Analysis of homologous recombination (HR) ef ficiency after transfection of the HDR-GFP reporter (Exp.2). A and C,analysis of extra-chromosomal HR ef ficiency in porcine fetal fibroblasts transfected with the HDR-GFP reporter, as determined by flow cytometry after electroporation with the CRISPR interference (CRISPRi) and Natronobacterium gregoryi Argonaute(NgAgo) interference (NgAgoi) systems. B and D, summary of HR ef ficiency in porcine fetal fibroblasts in the CRISPRi system and NgAgoi system. Data are mean±SE (n=3). Different letters mean significant difference at P<0.05 level. E, Western blot results for reduced KU70 and KU80 protein levels under corresponding CRISPRi and NgAgoi.
In the CRISPRi system, only some sgRNAs were able to significantly reduce the expression of related genes,such as sgRNA3 inPNKPand sgRNA1 and sgRNA3 inKU70, which may be related to whether the target sites are located within the core promoter region. According to a new study, Qiet al. (2013) found that targets located in the –35 bp region before the start codon could significantly inhibit gene expression, but when the target is in an adjacent area, the knockdown became less ef ficient. When targeting the promoter sequences, the silencing ef ficiency of the positive and negative strands was similar in the CRISPRi system (Bikardet al. 2013).In the NgAgoi system, the suppression of NHEJ pathway candidate genes was significantly reduced, and the positive and negative strand silencing ef ficiencies were similar, which is consistent with a previous study by Qiet al. (2016). Qiet al. (2016) found that NgAgo can bind to a target gene to block its transcription in zebra fish,thereby concluding that the gDNA/NgAgo system can clearly decrease the expression of these genes. However,Yeet al. (2017) found that mutants with an aspartate-toalanine substitution cleaved an RNA target as well as wild-type NgAgoin vitro, suggesting that these residues do not constitute an active site and that the RNase activity of the NgAgo mutants was responsible for the phenotype.It was subsequently con firmed that NgAgo may be a type of RNA interference that can target the mRNA transcription product (Yeet al. 2017). However, many studies have shown that Argonaute fromThermus thermophilusorPyrococcus furiosuscan catalyze the cleavage of DNA targetsin vitrowhen in the presence of complementary 5′p-ssDNA (Swartset al. 2014a, 2015). Therefore, we postulate that the NgAgoi system can downregulate target genes by binding to both mRNA and DNA, and, as such, it is more productive than the CRISPRi system, which only binds DNA targets. DNA-guided DNA cleavage using Argonaute-family proteins has been discussed in previous works (Shenget al. 2014; Swartset al. 2014b), but those findings are of limited practical utility due to the need for supraphysiological conditions, such as high temperature.Recently, researchers have reported that NgAgo could be used as a genome engineering tool for editing the human genome. This allows us to infer that gene expression was reduced because the corresponding gene sequence had been knocked out. However, our results by T7E1 assays showed that NgAgo cannot cleave endogenous target locus. This also seems to con firm speculation by Qiet al. (2016) that NgAgo can only interfere with the gene expression and cannot cut the genome itself.
Fig. 4 Effects of interference on key genes in the non-homologous end-joining (NHEJ) and homologous recombination (HR)pathways by knocking down KU70 or KU80 gene using CRISPR interference (CRISPRi) or Natronobacterium gregoryi Argonaute(NgAgo) interference (NgAgoi) systems (Exp.3). NC, negative control. Values are shown as mean±SE (n=3). Lowercase letters indicate different saliency (P<0.05) between different treatment groups.
Previous studies have shown that the suppression of key factors of NHEJ pathways, such asKU70,KU80,andLIG4, can significantly improve the ef ficiency of HR(Brittonet al. 2013; Chuet al. 2015). In our study, the knockdown ofKU70andKU80also significantly improved HR ef ficiency to 185 and 158%, respectively, in the CRISPRi system. Compared with previous studies, this study used the CRISPRi system to increase HR ef ficiency,indicating that this technology has a greater effect on target genes and is closer to the ideal conditions than traditional siRNA. However, the knockdown ofLIG4,PNKP, andNHEJ1did not improve the ef ficiency of HR,suggesting that these genes may compensate for other ones. DNA repair mechanisms include microhomologymediated end-joining (MMEJ), single-strand annealing(SSA), HR, and NHEJ (Mullinset al. 2015; Changet al.2017). In this way, interfering with the individual genes of a single pathway may not certainly affect the ef ficiency of HR (Changet al. 2017). In this study, it was interesting that the NgAgoi system could significantly knockdown NHEJ pathway factors, but could not increase HR ef ficiency. This negative result may be related to the stability and sustainability of the 5´-phosphorylated gDNA in the cells. The 5´-phosphorylated gDNA cannot be produced in the cells the same way as CRISPRi system,and it depends on extracellular synthesis. After a single import event, the 5´-phosphorylated gDNA in mammalian cells becomes degraded within hours (Shenget al. 2014;Swartset al. 2014a). Thus, we guessed the NgAgoi system can significantly downregulate gene expression,but its degradation state may not be sustained more than 96 h. In addition, CRISPRi and NgAgoi target sites are different; CRISPRi target sites are in front of the start codon, and NgAgoi is in exons, leading to the different effect on other factors in the DNA repair pathway, although their interference ef ficiency is close. As indicated by downstream gene expression, after CRISPRi interference,the cells showed a positive response, such as feedback regulation. We speculate that they either increasedLIG4andPNKPexpression or decreasedXRCC4expression.Changes in the expression of repair factors may be associated with CRISPRi interference in theKU70orKU80regulatory regions. After NgAgoi interference, the cells did not show a positive repair response in the expression of repair factors. In this way, the lack of feedback regulation may lead to significantly different HR ef ficiency. Thus,further studies are needed to better understand the feedback mechanism of the HR and NHEJ pathways. The activity of the NHEJ pathway is high and remains stable throughout the cell cycle, but the HR repair mechanism is only active from the late S phase to G2 phase (Linet al.2014). Changes in the cell cycle are dynamic processes.NgAgoi and CRISPRi may be inconsistently affected by the cell cycle. Finally, as given above, DNA repair mechanisms include MMEJ, HR, SSA, and NHEJ (Mullinset al. 2015; Changet al. 2017), thus different technologies may cause differences in the choice of repair methods.
5. Conclusion
CRISPR or NgAgo interference can suppress nonhomologous end-joining molecules, but only the inhibition ofKU70andKU80by CRISPRi, but not NgAgoi, enhanced homologous-recombination-repaired ef ficiency. This work also demonstrates that NgAgo showed no cleavage activity to target DNA. The downstream factors of CRISPRi appear to be more active than NgAgoi.
Acknowledgements
We appreciate the technical assistance of the National Engineering Research Center for Breeding Swine Industry,Guangdong Wens Foodstuff Group Co., Ltd., Yunfu, China.This work was supported by the National Science and Technology Major Project for Breeding of New Transgenic Organisms, China (2016ZX08006002) and the Guangdong Province “Flying Sail Program” Postdoctoral Foundation,China (2016).
Appendicesassociated with this paper can be available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
 Journal of Integrative Agriculture2019年2期
Journal of Integrative Agriculture2019年2期
- Journal of Integrative Agriculture的其它文章
- Digital mapping in agriculture and environment
- Maize production under risk: The simultaneous adoption of off-farm income diversification and agricultural credit to manage risk
- miR-34c inhibits proliferation and enhances apoptosis in immature porcine Sertoli cells by targeting the SMAD7 gene
- Protective roles of trehalose in Pleurotus pulmonarius during heat stress response
- Kiwifruit (Actinidia chinensis) R1R2R3-MYB transcription factor AcMYB3R enhances drought and salinity tolerance in Arabidopsis thaliana
- Arbuscular mycorrhizal fungi combined with exogenous calcium improves the growth of peanut (Arachis hypogaea L.) seedlings under continuous cropping