
运动介导细胞自噬调控先天性免疫的研究现状及展望
2019-02-07 12:26:54丁树哲
体育科学 2019年11期
戈 哲,丁树哲*
(1.华东师范大学“青少年健康评价与运动干预”教育部重点实验室,上海 200241;2.华东师范大学体育与健康学院,上海 200241)
细胞自噬是细胞维持正常生命活动的基本生理过程。日本科学家大隅良典因在细胞自噬机制研究中所做出的卓越贡献而获得2016年诺贝尔生理学或医学奖(Mizushima et al.,1998)。细胞自噬的过程主要是自噬体形成后与溶酶体融合形成自噬溶酶体,进而降解内容物,降解后的氨基酸等小分子物质再被细胞循环利用。细胞自噬的机制十分复杂,受众多信号分子的调控,其具体机制至今仍不清楚。先天性免疫与适应性免疫相对,它是抵抗病原体入侵的第一道防线,主要由皮肤、粘膜以及众多固有免疫细胞组成。目前,众多研究表明,细胞自噬也是先天性免疫防御系统的一部分(Choi et al.,2018)。有学者通过流行病学调查发现,运动强度与上呼吸道感染率之间存在着J形曲线的关系(Nieman,1994)。长期中等强度的耐力运动能够提高唾液免疫球蛋白A(IgA)水平,急性大强度运动后即刻血清IgG、IgM和IgA的浓度下降(方勇等,2010;Klentrou et al.,2002)。这些研究从流行病学和体液免疫的角度阐述了运动对先天性免疫的调控作用,但通过运动手段干预细胞自噬从而调控先天性免疫的研究目前尚无相关报道。笔者将探讨这一领域的研究现状,以待为今后的研究提供新的思路。
1 细胞自噬
哺乳动物自噬体的形成是一个复杂的过程。细胞自噬是由Unc-51样自噬激活激酶1(ULK1)复合物起始的。ULK1复合物的主要成员包括ULK1(酵母ATG1对应哺乳动物的同源物)、自噬相关蛋白13(ATG13)、FAK家族相互作用蛋白 200(FIP200)和 ATG101(Choi et al.,2018)。在自噬诱导过程中,ULK1复合物转移到自噬起始位点募集III类磷脂酰肌醇3激酶复合物I。III类磷脂酰肌醇3激酶复合物I包括液泡分选蛋白34(VPS34)、Becline1(酵母ATG6的哺乳动物同源蛋白)、VPS15和ATG14L。III类磷脂酰肌醇3激酶复合物I在自噬体形成位点生成3-磷酸磷脂酰肌醇(PI3P),即吞噬泡。PI3P再募集PI3P结合蛋白,例如,WD重复结构域磷酸化蛋白相互作用蛋白2(WI-PI2B)和双FYVE结构域蛋白1(DFCP1),之后再进入自噬体的延伸,最终形成完整的自噬体(Zachari et al.,2017)。自噬体的延伸主要依赖两套类泛素样连接过程,并且ATG12-ATG5与ATG3之间存在相互作用,刺激ATG3结合酶的活性,促进微管相关蛋白1轻链3(LC3)与磷脂酰乙醇胺(PE)结合。LC3-PE的集中,有利于形成自噬体膜,其在自噬体扩张过程中起着关键的作用(图1)(Ravikumar et al.,2010;Sakoh-Nakatogawa et al.,2013)。PE 是大多数细胞内膜的主要成分,有利于自噬体膜的形成。自噬体成熟后,通过与溶酶体融合形成自噬溶酶体。这个过程由III类磷脂酰肌醇3激酶复合物II介导,III类磷脂酰肌醇3激酶复合物II主要包括VPS34、Becline1、VPS15和紫外线抵抗相关基因蛋白(UVRAG)(Bento et al.,2016),形成自噬溶酶体后降解其中的内容物,为细胞提供能源物质,维持细胞的正常代谢功能。
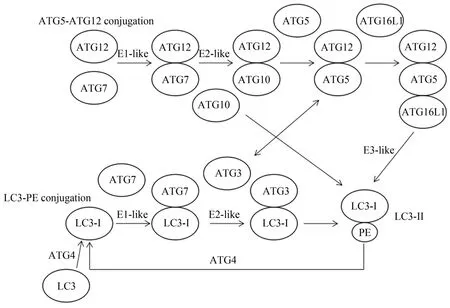
图1 自噬体形成的两套泛素样连接系统Figure 1.Two Ubiquitin-like Linking Systems in the Formation Process of Autophagosomes
2 细胞自噬与先天性免疫
细胞自噬是古老的先天性免疫反应之一。它是抵抗细菌、原生动物和外源性病毒的第一道防线,可以直接通过降解病原体的方式维持细胞的正常生命功能(Delgado et al.,2009),还可以通过以下多种途径诱导先天性免疫防御。
2.1 细胞自噬与干扰素
浆细胞样树突状细胞(pDC)在自噬抑制的情况下产生干扰素-α(IFN-α)的能力受到抑制,但病毒入侵pDC的过程并不受影响,且敲除Toll样受体7(TLR7)的pDC在单链RNA(ssRNA)病毒干预后不能产生IFN-α,以及敲除ATG5的pDC中I型干扰素生成大幅减少(Lee et al.,2007)。这表明TLR7对病毒的识别需要自噬过程的参与。同时,ssRNA病毒在pDC细胞中的识别完全依赖TLR7,且自噬功能的正常对TLR7信号通路至关重要。进一步探索发现,降低pDC细胞TLR7表达会导致自噬体形成减少和ATG7表达下降(Zhou et al.,2012)。这表明,pDC细胞很可能通过TLR7诱导细胞自噬进而产生IFN-α。并且,在病毒感染的树突状细胞(DC)中,存在自噬体的形成,细胞自噬与DC细胞免疫细胞因子的产生存在正相关关系(Morris et al.,2011)。
此外,自噬相关蛋白对先天性免疫信号通路也存在一定的调控作用。例如,交联的ATG5和ATG12与线粒体抗病毒蛋白(MAVS)和维甲酸诱导基因I(RIG-I)的CARD结构域结合,从而影响先天性免疫下游信号转导(Jounai et al.,2007)。并且,小鼠胚胎成纤维细胞敲除ATG5后可以增强I型干扰素的生成,同时细胞内受损线粒体增多、MAVS表达增多以及线粒体活性氧(ROS)生成增多(Tal et al.,2009),表明ATG5对RLR信号通路起着负性调节作用。mATG9抑制双链DNA(dsDNA)诱发的cGAS-干扰素基因刺激蛋白(STING)通路下游STING和TBK1的激活,降低I型干扰素的产生,而ATG5的表达则对TLR7通路产生I型干扰素起正向调控作用(Saitoh et al.,2010;West et al.,2015)。总体来看,自噬相关蛋白对RLR信号通路和cGAS-STING通路存在负性调控作用,而对TLR7信号通路则是正向调控作用,具体意义仍有待进一步研究。
2.2 SLRs受体
细胞自噬可以选择性地直接降解细胞内的病原体,主要通过SLRs来完成。其主要成员包括p62(又名SQS-TM1,一种泛素结合蛋白)、BRCA1基因相邻蛋白(NBR1)、核点蛋白52(NDP52)和自噬受体蛋白optineurin。它们均含有LC3相互作用区域(LIR)和泛素结合区域,从而引发自噬降解入侵的病原体(Deretic,2012)。入侵的细菌必须通过泛素化标记才能被SLRs受体识别。细菌被泛素标记的机制目前有两种:第1种是细菌通过胞吞作用或吞噬作用进入细胞,被核内体膜包裹,然后释放毒素破坏核内体膜,被破坏的核内体膜被泛素化标记,从而引发自噬,降解仍在核内体膜中心的细菌;第2种是细菌从破裂的核内体膜内逃逸,其表面蛋白直接被泛素标记,从而引发自噬降解细菌(Fujita et al.,2011),但具体是通过哪种机制仍需进一步研究。辛德毕斯病毒(SINV)、基孔肯雅病毒(Chikungunya)、单纯疱疹病毒1型(HSV-1)都能够被p62识别(Wileman,2013)。这表明病毒的识别机制与细菌类似,同样涉及泛素化标记作用。除此之外,先天性免疫分子TANK结合激酶1(TBK1)可以磷酸化optineurin的ser-177位点,提高其对LC3分子的亲和力,从而加强细胞自噬,清除胞浆的沙门氏菌(Wild et al.,2011)。IFN-γ还能直接调控自噬复合体来影响病毒的复制。例如,在巨噬细胞中,IFN-γ可以直接通过Atg5-Atg12/Atg16L1复合体抑制小鼠诺瓦克病毒(MNV)复制复合物的形成(Hwang et al.,2012)。这也说明,细胞可以通过自噬复合体直接抑制病毒的入侵,并不依赖于自噬后续的降解过程。
2.3 TLRs和RLRs
TLRs是模式识别受体(PPRs)家族成员之一,它可以直接识别细菌、病毒和寄生虫的病原相关分子模式(PAMPs)来诱发先天性免疫反应(Lee et al.,2007)。TLRs激活可以诱导自噬的发生,巨噬细胞中脂多糖(LPS)可以通过TLR4-β干扰素TIR结构域衔接蛋白(TRIF)途径诱导自噬的发生,敲低髓样分化因子88(MyD88)并不影响自噬体的形成(Xu et al.,2007)。除了TLR4,TLR家族其他成员也能在巨噬细胞中诱导自噬。并且,TLR信号能够加强MyD88、TRIF和Becline1之间的相互作用来诱导自噬(Shi et al.,2008)。这表明,MyD88与自噬的关系很密切,与先前研究存在矛盾,具体机制有待进一步研究。TLR7可以识别病毒,进而引发自噬来限制病毒的感染(Nakamoto et al.,2012)。进一步研究发现,TLR4诱导的自噬需要自噬关键分子Becline1,并且,Becline1的K63泛素化状态是自噬发生的关键(Shi et al.,2010)。此外,RLRs的激活也能诱导自噬的发生。研究发现,双链RNA模拟物聚肌胞苷酸Poly(I:C)能与黑色素瘤分化相关基因5(MDA5)受体结合,诱导自噬的发生(Tormo et al.,2009)。最新研究表明,RIG-1受体识别病毒RNA后,通过MAVS-肿瘤坏死因子受体关联因子6(TRAF6)信号诱导Becline1的K63多聚泛素化,进而诱导细胞自噬来抑制病毒的复制,这个过程不依赖干扰素的产生(Lee et al.,2018)。这些研究表明,RLRs可以直接通过激活细胞自噬,起到先天性免疫防御作用。
2.4 PKR
PKR存在于胞浆,其可以结合病毒双链RNA,然后磷酸化真核起始因子2α(eIF2α),进而诱导自噬的发生。然而,敲除PKR或者使eIF2α的ser-51去磷酸化突变后,自噬体形成出现障碍,并且HSV-1能够分泌神经毒性蛋白ICP34.5来抑制自噬,从而实现免疫逃逸(Talloczy et al.,2002)。Talloczy(2006)等进一步研究表明,PKR-eIF2α通路诱导的自噬,能够直接降解HSV-1,这也直接证明了细胞自噬的先天性免疫作用。
2.5 Nod样受体与CD46
Nod1和Nod2是NLR受体家族成员,它们能够结合入侵细菌的胞壁肽聚糖,从而诱导自噬的发生。这个过程不依赖受体相互作用蛋白2(RIP2)和核因子κB(NF-κB),而Nod1和Nod2诱导的下游炎症反应主要依赖于NF-κB信号和RIP2的募集。这表明了Nod1和Nod2诱导的自噬不依赖其下游NF-κB和RIP2引发的炎症反应。并且,Nod1和Nod2在这个过程中可以募集ATG16L1到细菌入侵细胞膜的位置上,进而诱导自噬来抵抗细菌的入侵(Travassos et al.,2010)。NLR受体家族成员之一NLRP4与Nod1和Nod2的作用相反。研究发现,其通过NACHT结构域(又称NOD结构域)与Becline1的保守区段进化保守结构域(ECD)结合,抑制自噬(Jounai et al.,2011)。
CD46是一种普遍存在于细胞膜表面的受体,它能够结合多种病原体诱导自噬。GOPC是一个支架蛋白,其有2个卷曲螺旋结构域(CC)和一个PDZ结构域(又名盘状同源区域)。CD46的1个剪切体CD46-Cyt-1与GOPC的PDZ结构域相互作用,GOPC的CC结构域与Becline1之间存在相互作用,从而与自噬体形成的复合物VPS34/Becline1结合,引发自噬。细胞表面CD46识别麻疹病毒和甲类链球菌后,通过CD46-Cyt-1/GOPC通路来激活自噬,从而起到控制感染的作用(Joubert et al.,2009)。
2.6 病毒对细胞自噬的抑制和利用
自噬在对抗外界病原体入侵方面起着重要的先天性免疫作用。但某些病毒为了对抗自噬,它们通过抑制自噬甚至利用自噬来维持自身的生存。例如,疱疹病毒属中的HSV-1、卡波济肉瘤相关疱疹病毒(KSHV)和鼠疱疹病毒68(MHV-68)编码的蛋白能够与Becline1结合,阻止自噬的发生(Rossman et al.,2009)。研究发现,禽流感病毒的M2蛋白可以阻断自噬体和溶酶体的融合过程(谷琳琳等,2017),从而阻止自噬降解的过程。还有研究表明,病毒也能利用自噬来增加自身的复制,乙型肝炎病毒(HBV)的小表面蛋白(SHBs)能够通过未折叠蛋白反应(UPR)来诱导自噬,UPR信号通路的阻断则导致SHBs不能诱发自噬。并且,发现抑制细胞自噬,HBV的数量减少,而诱导细胞自噬,则导致HBV复制增多(Li et al.,2011)。这表明HBV病毒的复制依赖自噬的激活,并且UPR能够诱导自噬的发生。
3 运动对细胞自噬的调控作用
3.1 运动通过AMPK/mTOR调控细胞自噬
运动过程中,肌细胞的强烈收缩导致ATP快速消耗,从而使得肌细胞内AMP/ATP比值升高,AMPK激活(Ferraro et al.,2014)。正常情况下,mTOR通过磷酸化ULK1 Ser757降低AMPK与ULK1的相互作用,从而抑制自噬。但在耐力运动干预情况下,ATP的不断消耗以及AMP的生成导致AMP/ATP比例升高,使AMPK被激活,然后通过磷酸化结节性硬化复合物2(TSC2)Thr-1227/Ser-1345和Raptor蛋白Ser722/Ser792,抑制mTORC1信号,进而降低ULK1 Ser757磷酸化。随后,AMPK与ULK1相互作用,使ULK1的Ser317/Ser777磷酸化,诱导自噬的发生(Kim et al.,2011;Sanchez et al.,2014)。AMPK与 ULK1相互作用,磷酸化激活ULK1位点,除了Ser317/Ser777外,还有Ser467、Ser555和 Ser637(Sanchez et al.,2014)。研究表明,6周的耐力运动能够降低小鼠肌肉mTOR表达的水平(苑红 等,2009),并使耐力运动后比目鱼肌AMPK Thr172、ULK1 Ser317和Ser555磷酸化增加、蛋白激酶B(AKT)Ser473和mTOR Ser2448磷酸化降低以及自噬标志物LC3-II表达增加(Pagano et al.,2014)。目前,大量研究表明,耐力运动能激活AMPK信号通路,充分说明耐力运动通过AMPK的激活抑制了mTOR,从而起到自噬诱导的作用。磷脂酰肌醇-3激酶(PI3K)/AKT/mTOR通路抑制细胞自噬,PI3K的激活导致磷脂酰肌醇2磷酸PI(4,5)P2和磷脂酰肌醇3磷酸PI(3,4,5)P3的生成,然后AKT的PH(pleckstrin homology)结构域与 PI(4,5)P2 和 PI(3,4,5)P3结合,从而使AKT的Thr308和Ser473磷酸化(Chen et al.,2014)。AKT使mTOR Ser2448磷酸化激活,进而抑制自噬(LoPiccolo et al.,2008)。众多研究表明,抗阻运动能够激活mTOR信号通路。有研究表明,大鼠经过8周的抗阻运动后,大脑皮质组织和海马组织的PI3K的P110亚基和P85亚基的表达水平以及AKT Thr308和Ser473磷酸化水平高于对照组(房国梁等,2016)。并且,小鼠在2周抗阻运动后,AKT Thr308、mTOR Ser2448与ULK1 Ser757的磷酸化水平升高以及p62增多,这表明抗阻运动通过PI3K/AKT/mTOR通路抑制细胞自噬(Steiner et al.,2015)。研究发现,年轻人和老年人进行一次急性抗阻训练3 h、6 h、24 h后,股外侧肌LC3B-II/LC3B-I比值下降(Fry et al.,2013)。肥胖人群进行8周的抗阻训练后,其肌肉AKT与下游mTOR活化,且mTOR分子的活化对于维持骨骼肌肌肉质量至关重要(Stuart et al.,2017)。上述研究表明,抗阻运动可以通过激活PI3K/AKT/mTOR通路降低自噬水平,从而降低肌肉蛋白质的降解速度来增加和维持肌肉质量。然而,有研究表明,9周抗阻训练后,大鼠肌肉LC3-II/LC3-I比值下降,自噬调节相关蛋白Becline1、ATG5等以及上游调控蛋白AMPK、磷酸化AMPK以及叉头框蛋白O3(FOXO3)表达增加,而且胰岛素样生长因子1(IGF-1)及其受体表达增加,下游AKT Ser473以及mTOR Ser2448磷酸化水平下降(Luo et al.,2013)。这又表明,抗阻运动后下游AKT以及mTOR的磷酸化变化与其他研究存在矛盾,原因有待进一步探讨。抗阻运动虽然诱导LC3B-II/LC3B-I的比值下降,但自噬相关蛋白表达升高,其具体机制也有待进一步探索。
3.2 运动通过FOXO3对细胞自噬的调控
AMPK还可以通过FOXO3激活自噬。研究发现,AMPK与FOXO3结合后磷酸化FOXO3 Ser413/Ser588,激活的FOXO3诱导自噬相关分子LC3-II、Gabarapl1和Becline1的表达,从而增强细胞自噬(Sanchez et al.,2012)。AKT则通过FOXO3抑制细胞自噬,还可以通过直接磷酸化FOXO3 Thr32/Ser253和间接磷酸化Ser315,使其定位在胞浆中。然而,细胞在加PI3K抑制剂LY 294002处理后,FOXO3则转移到细胞核中。FOXO3的Thr32/Ser315/Ser253突变后,其定位于细胞核中,并且胞浆的FOXO3 Thr32/Ser253位点磷酸化后与14-3-3蛋白存在相互作用(Brunet et al.,1999)。这表明,14-3-3可以通过结合磷酸化的FOXO3将其留在胞浆中,FOXO3的Thr32/Ser315/Ser253去磷酸化是其发挥转录因子功能的关键。还有研究表明,AKT可以磷酸化FOXO3 Thr 318/321,而AMPK激活降低FOXO3 Thr 318/321的磷酸化,诱导FOXO3的核转位增强细胞自噬(Tong et al.,2009)。这表明,AKT抑制FOXO3活性通过磷酸化Thr32/Ser315/Ser253和Thr 318/321,而AMPK激活FOXO3则通过磷酸化Ser413/Ser588。然而,调控FOXO3的功能是否还存在其他位点,有待进一步研究。研究表明,耐力运动后,小鼠比目鱼肌中AMPK Thr172激活,降低FOXO3 Thr32/Ser253的磷酸化(Pagano et al.,2014)。这充分说明AMPK的激活可以诱导FOXO3的去磷酸化,从而诱导细胞自噬。有研究表明,抗阻运动可以诱导FOXO3 Ser253磷酸化(Ato et al.,2017)。由此可知,抗阻运动可以通过激活AKT来抑制FOXO3的活性,抑制细胞自噬,而耐力运动可以通过AMPK激活FOXO3,激活细胞自噬。
3.3 运动通过Bcl-2/Becline1复合体对细胞自噬的调控
B淋巴细胞瘤-2(Bcl-2)是一个抗凋亡和自噬的蛋白。在正常情况下,它与Becline1的BH3结构域结合,从而抑制自噬的发生。耐力运动能够导致Bcl-2与Becline1解离,从而引发细胞自噬。然而,Bcl-2 3个磷酸化位点Thr69/Ser70/Ser84突变后,Bcl-2与Becline1相互作用并未出现分离,但是耐力运动导致的细胞自噬被抑制(He et al.,2012)。这表明,运动通过Bcl-2的这3个位点的磷酸化来诱导细胞自噬。除此之外,DAPK可以磷酸化Becline1 BH3结构域的Thr119位点,从而使Becline1与Bcl-XL分离,激活自噬(Zalckvar et al.,2009)。研究发现,激活JNK1也能使Bcl-2磷酸化,从而使Bcl-2与Becline1分离来激活自噬(Wei et al.,2008)。HMGB1对细胞自噬存在一定的调控作用,它是高度保守的细胞核蛋白。然而,氧化应激状态下,细胞内活性氧ROS的产生导致HMGB1转位到胞浆。HMGB1与Becline1相互作用,使Becline1与Bcl-2解离,从而激活自噬。胞浆的HMGB1还能通过磷酸化激活细胞外调节蛋白激酶(ERK1/2),使Bcl-2磷酸化,从而引发自噬(Tang et al.,2010)。这表明,运动很可能通过产生ROS,使HMGB1发生转位来引发自噬,具体机制仍有待进一步研究。并且,HMGB1的106位半胱氨酸突变后,胞浆中与Becline1结合的HMGB1增多,促进自噬。然而,HMGB1的C23和C45位点突变后,则不能与Becline1结合,失去其自噬诱导功能(Tang et al.,2010)。这表明,HMGB1分子内C23与C45分子内的二硫键的形成,对于结合Becline1引发自噬是必需的,而C106位的半胱氨酸是HMGB1细胞核定位的关键。除此之外,Becline1的K63多聚泛素化状态也是自噬激活的关键(Lee et al.,2018;Shi et al.,2010)。
4 运动、细胞自噬及先天性免疫的关系
中等强度的运动可降低上呼吸道的感染率,增强先天性免疫。大强度运动则反之,这早已被学界所公认。这也表明,不同强度的运动方式对先天性免疫存在不同的调控。目前,国内外学者在运动对先天性免疫的研究多集中在免疫细胞。例如,中等强度耐力运动后,小鼠巨噬细胞吞噬能力增强(姚毓才等,1994),且长期抗阻运动可以提高老年女性安静时NK细胞的活性(Mcfarlin et al.,2005),增强先天性免疫。然而,也有研究表明,渐进抗阻运动对先天性免疫没有影响(崔思松,1998)。相反,高强度频率的抗阻运动通过降低NK细胞活性进而降低先天性免疫(Kawada et al.,2010),这可能与抗阻运动的方式、强度及频率有关。然而,不同运动方式改变这些免疫细胞功能的机制仍不清楚。此外,运动对先天性免疫受体的研究多集中在TLR4。TLR4是TLRs受体之一,可以识别细菌LPS,产生干扰素和炎症因子,来引发先天性免疫反应(Lu et al.,2008)。研究表明,有规律的耐力运动可下调脑中风大鼠脑组织中TLR4的水平(Zwagerman et al.,2010),并且,急性有氧运动和长期的抗阻运动能够下调单核细胞表面TLR4的表达,从而减缓单核细胞炎症反应能力,降低机体炎症反应(Gleeson et al.,2006)。然而也有研究表明,急性大强度耐力运动1 h后,大鼠心脏组织TLR4mRNA 水平上调(Cristi-Montero et al.,2012)。这表明,低强度的运动降低TLR4的表达,大强度的运动增加TLR4的水平,这可能与组织炎症水平有关。
细胞自噬可以起到先天性免疫防御作用,不同的运动方式对细胞自噬和先天性免疫存在不同的调控机制。耐力运动通过AMPK/ULK1信号AMPK/FOXO3以及Becline1的激活来诱导细胞自噬的发生。并且,Becline的激活受到DAPK、JNK1、HMGB1以及自身K63泛素化状态的影响。而抗阻运动则通过激活AKT/mTOR信号和AKT/FOXO3信号抑制自噬的发生。然而,抗阻运动是否激活自噬仍然存在一定的争议。有研究表明,抗阻运动能够增加VPS34的活性(Mackenzie et al.,2007)。VPS34与自噬体的形成至关重要,推测其活性的增加可能与降解受损的肌纤维细胞器有关。并且,未受训练过的人进行一次抗阻运动48 h后,其骨骼肌LC3-II水平增加(Hentila et al.,2018),提示细胞自噬作用增强。鉴于细胞自噬的先天性免疫防御作用,运动所诱导的细胞自噬很可能联系着先天性免疫的增强。运动强度与TLR4的表达呈正相关关系,这与中低强度的运动增强先天性免疫的结果相反。由于TLRs能够诱导自噬的发生,不难推测长期规律的耐力运动可能并非通过TLR4诱导自噬的发生来增强先天性免疫。同时,目前尚无运动对其他TLRs、RLRs等免疫受体的报道,而这些免疫受体均能引发细胞自噬来消灭病原体。该问题亟需进一步研究。细胞过度自噬可能导致免疫功能的下降。过度的耐力训练导致大鼠肌肉自噬相关基因ATG7、Becline1、LC3-II和FOXO3表达大幅增加,血液淋巴细胞百分比下降(Feng et al.,2011),推测其与肌肉损伤以及体内炎症水平的升高有关,但具体机制目前尚不清楚,有待进一步研究。另外,运动所诱导的细胞自噬不仅仅局限于骨骼肌,还能影响其他细胞及组织。例如,耐力运动可以在胰岛β细胞、肝脏和脂肪组织中通过Bcl-2/Becline1复合物诱导细胞自噬(He et al.,2012),但目前尚未有研究证明运动可以诱导机体免疫细胞及非免疫细胞的细胞自噬来限制和消灭病原体,这个问题未来亟需研究。综上所述,细胞自噬的过程极其复杂,众多分子以及SLRs等免疫受体均参与细胞自噬。并且,不同运动方式又能够通过多种方式对自噬进行调控(图2)。
5 小结与展望
耐力运动和抗阻运动通过不同的方式对细胞自噬存在一定的调控作用,而细胞自噬与先天性免疫密切相关。耐力运动很可能通过细胞自噬的方式增强先天性免疫。然而,某些病毒也可进化出相应的机制来抵抗自噬,甚至利用自噬来增加自身的复制。目前,仍有以下问题亟需研究:1)抗阻运动对细胞自噬以及先天性免疫的影响亟需进一步探索;2)目前,运动对自噬的研究集中在肌细胞,那么运动能否通过诱导肌细胞或其他细胞自噬直接消灭入侵的病原体?3)SLRs、TLR、PKR、Nod1/Nod2和CD46可以通过识别病原体从而诱导细胞自噬的发生,DAPK和JNK1的激活、HMGB1的胞浆定位和Becline1的K63多聚泛素化状态同样也是调控自噬激活的关键因素。目前,运动对细胞自噬的影响集中在AMPK/mTOR等传统信号分子上,而运动能否通过这些分子调控自噬进而影响先天性免疫仍是一个疑问,有待进一步研究。

图2 不同运动方式、相关分子及免疫受体通过细胞自噬对先天性免疫的调控作用Figure 2.Regulation of Innate Immunity through Autophagy by Different Exercise Types,Molecules and Immune Receptors
猜你喜欢
汽车与运动(2023年11期)2023-03-04 08:03:26
天津医科大学学报(2019年6期)2019-08-13 07:04:42
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
车迷(2017年10期)2018-01-18 02:10:57
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
遗传(2014年3期)2014-02-28 20:59:01
军事体育学报(2014年3期)2014-02-27 16:00:12
