
酶法合成环肽研究进展
2019-01-29 10:26程孝中周志昉吴志猛
食品与发酵工业 2019年1期
程孝中,周志昉,吴志猛
(江南大学,糖化学与生物技术教育部重点实验室,江苏 无锡,214122)
环肽化合物在自然界中普遍存在,与线性肽相比,它可以形成刚性构象,提高了对蛋白酶降解的抵抗能力;某些环肽还具有独特的细胞膜穿透能力;另外,小分子环肽对某些蛋白受体具有较高的亲和力和选择性[1]。环肽因其优良的生物活性及构象特征,在生物医药领域得到广泛关注。自从1944年苏联科学家发现了第1个环肽短杆菌肽(gramicidin S)[2],这种治疗伤口感染的环肽挽救了无数生命。至今为止,研究者从动植物、微生物中均发现了环肽,例如,抗生素类的短杆菌酪肽A、乳酸链球菌肽;免疫抑制剂类的环孢菌素A;抗癌抗病毒因子类的万古霉素等[3]。
目前,环肽的合成方法主要有:(1)化学合成法。使用该法合成环肽需要侧链全保护的线性多肽,当环肽氨基酸数量增多,由于熵的壁垒增加,导致合成效率降低。近年来又发展了很多位点特异性的反应用于环肽合成,如化学自动连接反应(native chemical ligation, NCL)[4],点击化学(click chemistry)[5],酮酸-羟胺偶联法(ketoacid-hydroxylamine ligation)[6],氮丙啶醛法(aziridine aldehydes-mediated macrocyclization)[7]等。(2)生物合成法。基于生物体内转肽机制发展起来的一些方法,例如利用内含肽自我剪接的表达蛋白连接(expressed protein ligation,EPL)[8]、蛋白反式剪切(protein trans-splicing,PTS)[9],以及依赖体外的翻译系统的遗传密码重编程(genetic code reprogramming),这类方法主要针对大分子蛋白的环化,一般都要进行基因表达获得线性底物。
酶催化的环肽合成是近年来兴起的一种新型环肽合成方法,该法具有反应条件温和、特异性强、合成效率高等优点,另外酶催化的环肽合成还可以在肽链中引入非天然氨基酸或非氨基酸结构,进而大大丰富了环肽结构。本文就近几年来发展的酶法合成环肽进行综述,本文涉及到的环肽包括均环肽和杂环肽,这些环肽的连接方式主要有首尾连接、侧链与首或尾连接以及侧链连接。
1 介导环肽合成的酶
1.1 分选酶Sortase A
分选酶A(Sortase A)是从金黄色葡萄球菌(Staphylococcusaureus)分离出来的1种转肽酶,能特异地识别肽C端的保守序列LPXTG(Leu-Pro-X-Thr-Gly;X代表任1种氨基酸),并从苏氨酸和甘氨酸之间使肽键断裂,形成酰基酶-底物复合物,N端带有多聚甘氨酸的亲核基团进攻复合物,最终形成连接产物(图1-a)[10]。该酶基因工程表达产率较高(232 mg/L),易于获取,已广泛地用于蛋白连接或蛋白标记[11]、细胞表面的蛋白标记[12]和蛋白环化[13]。
WU等系统性地探索了分选酶A催化多肽底物环化时肽链的最适长度,发现线性底物肽最短为19个氨基酸(包括信号序列LPXTG和GG)时,反应产物主要是单环产物,化合产率达到80%;当线性肽核心序列小于17个氨基酸(包括信号序列LPXTG和GG)时,环化单环产物仅为6%,主要形成二聚体和三聚体环肽,而且底物浓度增大,二聚体环肽和三聚体环肽增多,单环产物相应减少[14]。这种规律还和底物肽序列有关,在利用分选酶A合成RGD环肽过程中,当底物肽为16个氨基酸时(包括信号序列LPXTG和GG),主要产物仍是单环肽[15]。分选酶A通常在大环肽合成方面产率更好,在合成唾液肽组胺素(环肽为38个氨基酸)产率能够达到90%[16],可能是因为底物肽链越长,N端多聚甘氨酸在空间上更容易接近LPXTG。另外,分选酶A强大的环化能力还可以用来合成复杂的二硫键环肽(胰蛋白酶抑制剂SFT-1、芋螺毒素Vc1.1和抗病毒肽kB1),即使产物中引入了信号序列(LPXTG),并不影响其生物活性[17],例如利用该酶对人干扰素和生长激素的环化[18-19]。ZHANG等拓展了分选酶A的应用范围,在环肽SFT-1非保守区域嫁接不同功能的小肽基序,赋予SFT-1更多的生物功能,形成特异的配体嫁接环肽可以调节蛋白质之间的相互作用[20]。
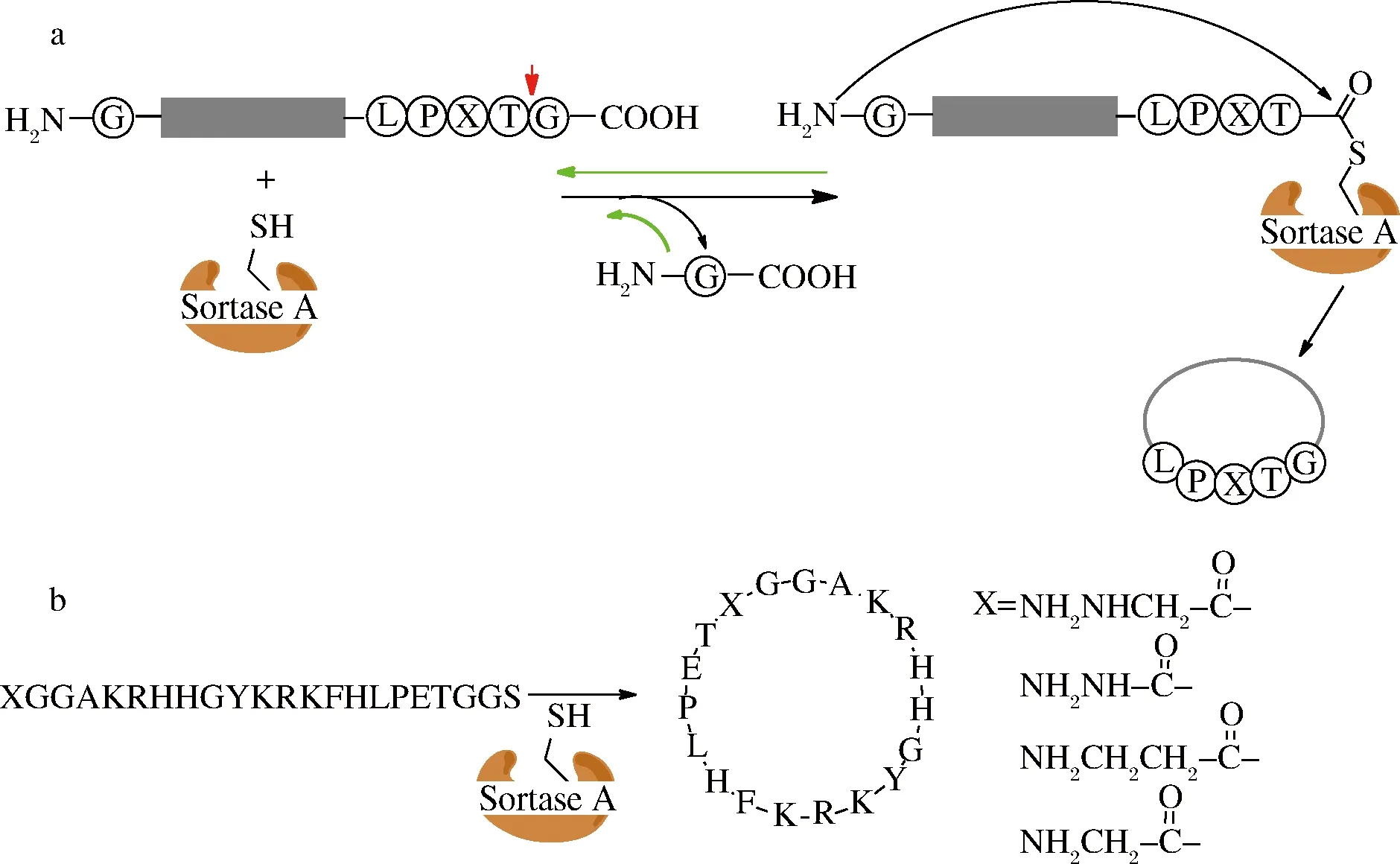
图1 分选酶A介导的环肽合成(a);N端不同的甘氨酸模拟物抑制分选酶A环肽合成(b)中的可逆反应
Fig.1 Sortase A-mediated cyclization of peptides(a),glycine mimics at N terminus inhibit reversible reactions of sortase A -mediated cyclization of peptides(b)
分选酶A催化效率在同类酶中较低(与底物的摩尔比例为0.1~1.0),因此反应需要更长的时间(>20 h)和更高的酶量。另外,分选酶A催化反应是一个可逆反应。为了使反应平衡朝目的产物方向进行,可以加大反应底物的量或者及时清除反应中的副产物[21]。PLOEGH课题组将分选序列(LPXTG)C端的甘氨酸用甲酯替代,转肽反应形成连接产物后,释放出甲醇而不是甘氨酸亲核基团,阻止逆反应进行[22],但这种反应的产率并不高,而用羟基乙酸替代甘氨酸后的反应速率更高[23]。LIU对甘氨酸亲核试剂进行改进,利用酰肼代替甘氨酸作为亲核试剂进攻酶-底物复合物形成一种非天然的环肽产物[24],本课题组利用这种策略合成了非天然的环肽类似物抗菌肽P-113,产率达到76%~93%[25](图1-b)。
1.2 肽连接酶Butelase-1
蝶豆兰花肽连接酶-1(Bunga telang plus ligase,Butelase-1)是一种天冬酰胺内切蛋白酶(asparaginylendoproteases,AEP),参与热带药用植物蝶豆中大环肽合成[26]。2014年由TAM课题组首次从碟豆中分离出来,能识别肽链C端三肽序列D/N-HV,并从组氨酸前面进行肽键的断裂,释放HV二肽序列并形成酰基酶底物复合物,随后的N端氨基酸作为亲核基团进攻复合物重新形成肽键(图2),产物中连接位点只留下D/N印迹(分选酶A为LPXTG)[27]。蝶豆兰花肽连接酶-1对底物N端第1,2位氨基酸有一定要求,第1位氨基酸可以为除Pro、Asp和Glu以外的任何天然氨基酸,第2位为疏水性氨基酸Ile、Leu、Val以及Cys[28]。

图2 蝶豆兰花肽连接酶-1介导环肽合成
Fig.2 Butelase-1-mediated cyclization of peptides
蝶豆兰花肽连接酶-1催化效率很高(0.005摩尔当量),可以在很短时间内环化10个氨基酸或者更长的环肽(70个氨基酸)生成[29-30]。这种酶的催化反应水解产物较少,更倾向环化产物形成,例如,合成29个氨基酸的环肽kalata B1产率达到95%以上,底物的耐受性较好,除C端D/N氨基酸以外,其余的氨基酸都可以为D型氨基酸[28]。
虽然蝶豆兰花肽连接酶-1催化效率高,但是和分选酶A催化的反应一样,该反应也是可逆反应,反应过程中需要大量底物促使反应进行。另外,蝶豆兰花肽连接酶-1目前没能实现基因工程表达,即使添加GST或MBP促溶标签也无法进行原核表达,这限制了蝶豆兰花肽连接酶-1应用前景。HARIIS课题组通过突变获得了1种AEP突变体OaAEP1,在进化地位上与蝶豆兰花肽连接酶-1相关,功能类似,可以实现在大肠杆菌中进行表达,但产率较低(<2 mg/L),催化效率也远远低于蝶豆兰花肽连接酶-1[31-32]。因此,实现蝶豆兰花肽连接酶-1基因工程表达,提高其产量仍是一个重要课题。
1.3 枯草杆菌蛋白酶(subtillisin)突变体
早在1997年,WELLS课题组利用基因工程技术从解淀粉芽孢杆菌得到了一种枯草杆菌蛋白酶突变体(S221C,P225A),命名为subtiligase,并初次实现了该酶的环肽合成,产率为36%~85%,当环肽大于14个氨基酸时,主要是环肽产物,小于14个氨基酸的产物主要以水解产物和二聚体为主[33]。但该酶的稳定性较差,反应过程易生成水解产物。TOPLAK课题组随后通过基因工程方法表达了另1种来源于枯草芽孢杆菌的枯草杆菌蛋白酶突变体肽连接酶(peptiligase, S221C,P216A),这种蛋白的表达产率达到500 mg/L以上,而且酶稳定性和催化能力都得到了极大改善[34]。这种突变体连接酶并不依赖于钙离子,酶的巯基首先与C末端的甲酰胺甲基酯(carboxamidomethyl-ester,oCam)反应形成复合物,N端氨基作为亲核试剂进攻酶复合物并形成产物(图3),这和分选酶A的催化机理类似[34]。催化形成的环肽产物与水解产物之比(S/H)与底物肽长度有关,形成13个氨基酸产物时的环肽产物与水解产物之比(S/H)大于100,线性肽底物1 h内的转化率>99%,而转化为水解产物不足1%。并且该反应可以允许有机溶剂(50% DMSO和DMF)和变性剂(2 mol/L尿素或者盐酸胍)存在,这可以使那些水溶性不好的多肽实现环化[34],扩大了酶的使用范围。
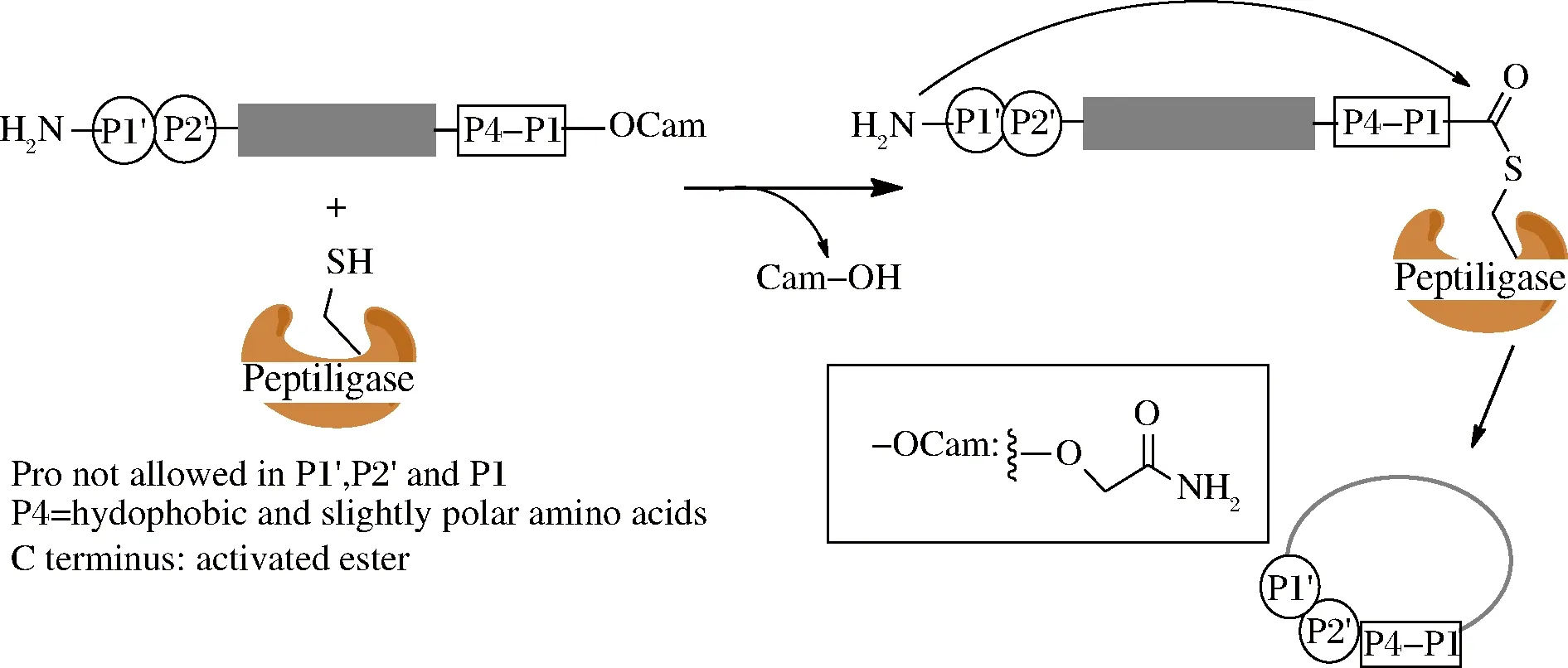
图3 枯草杆菌蛋白酶突变体肽连接酶介导环肽合成
Fig.3 Peptiligase-mediated cyclization of peptides
枯草杆菌蛋白酶突变体肽连接酶(peptiligase)同样需要识别序列,这个序列包括C末端的4个氨基酸(P1~P4)以及N末端的2个氨基酸(P1’和P2’),C末端需要形成活化酯结构,P1’氨基酸主要为Ser、Gly和Ala,P1’和P2’不能为Pro,另外,P4位适合疏水性氨基酸(图3)。而另1种枯草杆菌蛋白酶突变体肽连接酶(omniligase-1)底物宽泛性更好,该酶识别序列的P1’可以为除了Pro以外的任何氨基酸[35]。而且能够实现最小环化的氨基酸数量为11个,当环肽氨基酸数量大于12个氨基酸,可以获得90%产率,并实现了克级别环肽的合成。另外该酶还可以合成复杂环肽,比如34个氨基酸的二硫键环肽MCoTⅠ-Ⅱ,在产物中引入活性位点序列、D型氨基酸以及侧链异肽键形成都能高效的实现。
和分选酶A,蝶豆兰花肽连接酶-1相比,枯草杆菌蛋白酶突变体肽连接酶催化效率更高(<0.0003摩尔当量),由于该连接酶并不是转肽酶,是个非可逆反应,这样反应水解产物量很低,但需要在底物肽C末端引入活化酯结构。
1.4 硫酯酶(thioesterase)
自然界中生物合成的环肽可以分为核糖体环肽和非核糖体环肽,非核糖体环肽的合成是由非核糖体肽合成酶(nonribosomal peptide synthetases,NRPSS)完成,这种酶是目前发现的最大的酶复合物,由于没有核糖体帮助,非核糖体环肽合成依赖于这种大的酶复合物完成氨基酸运载、肽键形成、多肽转移与释放等多种功能[36]。酶复合物中最为关键的结构域为硫酯酶结构域(thioesterase domain,TE-domain),该结构域具有环化、终止反应和释放产物功能,含有保守催化三肽序列(SHD),其中Ser侧链羟基进攻底物肽C端硫酯结构(常用的有SNAC)中的羰基形成肽-酰中间产物,中间产物可以被水解成线性肽,也可以被分子内的亲核基团(如氨基或者羟基)攻击形成环化产物(图4),亲核基团可以来源于N端(如氨基)或者侧链[37]。
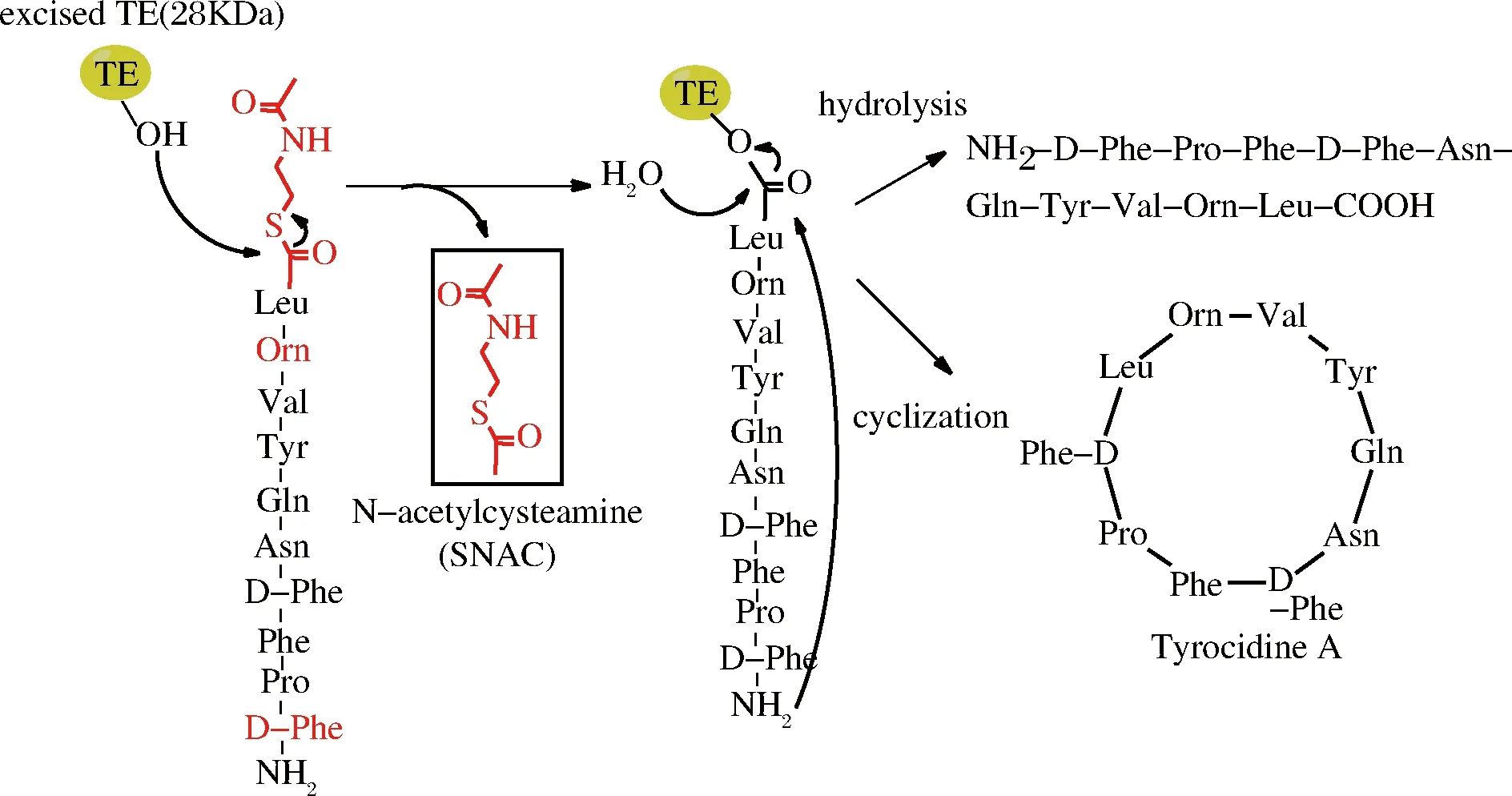
图4 TE结构域介导的环肽合成(以合成短杆菌酪肽为例)
Fig.4 TE-mediated cyclization of peptides
早在2000年,WALSH课题组从短杆菌Bacillusbrevis中克隆了合成短杆菌酪肽TE结构域TycC TE(tyrocidine TE,28 kDa),在体外成功合成了环肽短杆菌肽S和短杆菌酪肽[36],随后他们又分别从Bacillusbrevis,Bacillussubtilis中克隆了合成短杆菌肽和脂肽的TE结构域GrsB TE和SrfA TE,都能实现体外合成相应环肽[38]。TRAUGER等探索了TycC TE对底物的专一性,N-端的D-Phe以及序列中9位稀有氨基酸Orn对环化影响较大,鸟氨酸能够使底物肽形成β结构,这样易于与酶发生相互作用,因此,线性肽N端和C端氨基酸种类以及底物的二级结构对环肽合成具有重要作用[36]。
为了拓展TycC TE在合成环肽方面的应用,WALSH课题组将固相合成与酶法催化结合起来,利用PEGA树脂作为载体,在96孔装置里一步法进行多肽固相合成与酶法环化,合成了短杆菌酪肽库用于筛选活性更好的抗菌肽,环化产物与线性水解肽比例约为0~48.1,由于水解产物,最终环化产率较低[39]。WU等利用同样策略合成短杆菌肽S,固相载体改为TentaGel树脂,TE结构域为GrsB TE,但树脂上只有28%的线性肽进行了环化,即使增加反应温度以及酶量,都无法提高转化率[40],这可能与酶没法进入固相载体与底物肽进行充分的接触有关,需要对酶的大小尺寸进行基因工程改进,或者改变固相载体。
硫酯酶突变体在体外合成环肽主要模拟生物体内合成抗菌肽的机理,特定的TE结构域往往只能合成特定的环肽及其类似物(如合成短杆菌肽、短杆菌酪肽和脂环肽及其类似物),对其他序列的适应性并不清楚,序列中需要含有大量的非天然氨基酸和稀有氨基酸以形成酶的最适底物构象,难以推广至其他环肽合成。目前为止,大量的水解产物问题并没有得到很好的解决。
1.5 PatG相关结构域
海洋蓝藻环肽(cyanobactins)是由海洋生物体内合成的核糖体环肽,具有抗肿瘤活性。合成此类环肽也是由一系列的酶协调完成,2009年,SCHMIDT等从珊瑚礁中克隆了合成该海洋蓝藻环肽的环化功能的结构域(PatG),并在体外合成了该环肽及其类似物[41]。这种酶催化环肽合成机理类似于硫酯酶,也会形成水解线性肽副产物,区别在于它并不需要引入活化酯结构,因此,底物线性肽可以直接利用固相合成技术制备[42]。NAISMITH课题组随后从Prochloronsp. 中克隆了合成小盘酰氨(patellamide)的环化结构域(PatGmac)[43],这类酶首先识别底物肽C端信号序列AYG(E),酶上的羟基进攻A前面的肽键并形成酰基酶复合物,同时释放AYG(E)短肽,N端的氨基作为亲电基团进攻酯结构中的羰基完成环化[44]。AYG(E)中丙氨酸前面(P1位)的氨基酸对环肽合成至关重要,一般都是脯氨酸或者噻唑啉结构(如图5中Tzl),这种结构能够使酶与底物形成最佳结合构象[45]。另一种从海洋蓝藻中发现的OscGmac对P1位的宽泛性更好,利用Ser和Cys模拟的脯氨酸结构都可以作为环化底物,包括底物中引入D型氨基酸,对6~11个氨基酸长度的底物环化产率较好[46]。这种酶和硫酯酶一样,都是从生物体内合成特定环肽的酶复合体中分离得到的环化结构域,对底物序列是有一定要求,大范围的改变序列会导致环化失败,而且正常环化产率并不高(32%~58%)[47]。
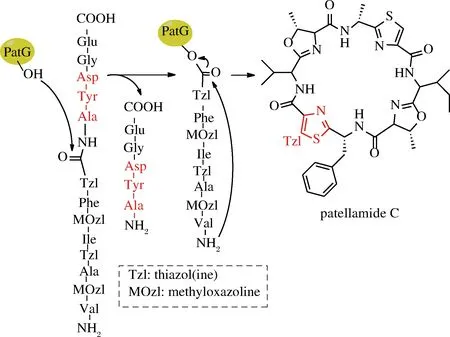
图5 PatG介导环肽合成(以合成patellamide C为例)
Fig.5 PatG-mediated cyclization of peptides
1.6 谷胱甘肽S-转移酶(glutathione S-transferase,GST)
PENTELUTE课题组首次报道了利用GST偶联原理来合成大环肽,这种环化方式主要是侧链之间的连接。他们将多肽N端接入GST识别标签ECG三肽序列,C端通过化学合成引入氟苯亲电试剂,在GST作用下氟苯与ECG中的半胱氨酸巯基进行共价连接(图6)[48]。对比不同氟苯亲电试剂发现十氟联苯效果最好,反应速度更快,5 min能够完成90%以上的环化。环肽链在一定长度范围内(14~24个氨基酸),随着肽链增长,环化产率也相应增大(93%~96%),肽链为40个氨基酸时,可以获得70%产率。对于其他的转肽酶,底物肽浓度增加会导致多聚体副产物生成,而这种环化当底物肽浓度达到10 mmol/L,而酶浓度只需要摩尔分数为0.1时,就检测不到多聚体副产物。另外,GST适应的温度范围比较广(4~60 ℃),水相中允许添加有机溶剂可以达到20%。但和NCL方法类似,这种环化方法仍需要引入巯基以及活性较好的亲电基团[48]。
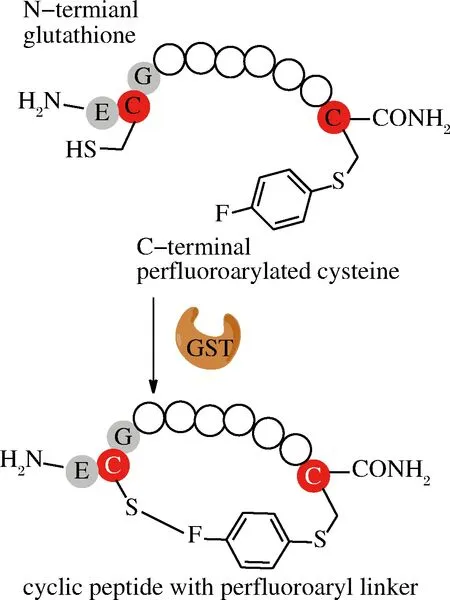
图6 GST介导环肽合成
Fig.6 GST-mediated cyclization of peptides
1.7 谷氨酰胺转氨酶(transglutaminase,TGase)
在自然界中,TGase主要催化蛋白质或者多肽中谷氨酰胺上的酰胺与其他伯胺之间的酰基转移反应,参与蛋白质与蛋白质或者小分子之间的交联形成异肽键。在食品工业上,TGase经常用来进行肉质酥松,也可以对大分子进行小分子偶联修饰,例如,将亲和素、荧光基团等偶联至抗体上[49]。但是利用TGase进化环肽合成报道较少。
TOUATI等利用来源于微生物链霉菌(Streptomycesmobaraensis)的谷氨酰胺转氨酶(MTGase,38 kDa)尝试了血浆激肽释放酶抑制二环肽合成(图7)[50]。

图7 MTGase介导的环肽合成
Fig.7 MTGase -mediated cyclization of peptides
N端设计成酶的识别序列WALQRPH,其中Q作为酰基供体,C端引入了KS序列,K作为酰基受体,中间使用多聚甘氨酸链作为连接。这种识别序列只能置于N端,易于酰基供体与受体在空间上靠近。信号序列截短实验发现酰基供体Q前面的2个氨基酸对环化非常重要,在底物浓度较高情况下,同样会出现二聚体副产物。
2 结论与展望
基于环肽复杂的拓扑结构和独特的生物活性,环肽逐渐成为研究热点,有可能解决某些大蛋白无法成药的困境。酶法和多肽固相合成技术相结合可以合成结构复杂、种类多样的环肽结构,为药物筛选提供平台。为了降低酶法合成成本,实现工业化级别合成,酶的获取成为关键因素,目前,有些酶实现了商品化,例如,枯草杆菌蛋白酶突变体肽连接酶(omniligase-1)已经开发为试剂盒,可以进行克级别的环肽制备,即使有些来源于植物的环肽合成酶暂时还没法实现基因工程表达,但相信这些问题都会得到解决。另外,生物体内非核糖体环肽合成酶可以合成多样化的环肽,这为研究者提供了一个环化酶的筛选平台,利用分子生物学技术筛选定制酶也会成为一个新的研究方向。借助于计算机工具以及多肽发现平台,酶法合成可以作为化学合成策略很好的补充,加速未来环肽疗法的发展。
猜你喜欢
城市道桥与防洪(2022年3期)2022-05-08
安全与环境工程(2021年2期)2021-04-02
农业科技通讯(2021年1期)2021-03-06
教育周报·教育论坛(2020年3期)2020-10-21
科学(2020年2期)2020-08-24
煤炭加工与综合利用(2020年6期)2020-07-17
科技资讯(2018年16期)2018-10-26
科技资讯(2017年12期)2017-06-09
上海农业学报(2017年3期)2017-04-10
天津医科大学学报(2015年2期)2015-12-22
