
磁性分子印迹聚合物提取-超高效液相色谱-串联质谱法测定乳及乳制品中的4 种伪蛋白
2019-01-28 08:06:50王象欣陈美君姜毓君满朝新
食品科学 2019年2期
单 艺,王象欣,陈美君,姜毓君,3,满朝新,3,马 微
(1.东北农业大学黑龙江省绿色食品科学研究院,黑龙江 哈尔滨 150028;2.东北农业大学食品学院,乳品科学教育部重点实验室,黑龙江 哈尔滨 150030;3.食品安全和营养协同创新中心,黑龙江 哈尔滨 150028;4.东宁出入境检验检疫局,黑龙江 东宁 157200)
三聚氰胺、环丙氨嗪、双氰胺和缩二脲这4 种含氮量较高且性质稳定、价格低廉的化合物被称作伪蛋白。添加到乳制品中使得蛋白质的含量远高于其实际含量,从而提高乳制品的附加值。同时,这类伪蛋白可作为动植物饲料,被广泛的应用于反刍动物饲养以及草场肥料等方面,也大大增加了其进入乳制品中的风险,严重危害人类健康[1-3],阻碍企业乃至行业的生存发展[4]。目前食品安全国家标准规定的蛋白质测定方法为凯氏定氮法,该法通过测定氮元素的含量间接对食品中蛋白质进行定量,而不是测定蛋白质的真实值。
根据卫生部等5个部门关于三聚氰胺在食品中的限量值的公告(2011年第10号),我国对婴幼儿配方粉中三聚氰胺的最高残留限量为1 mg/kg。美国和欧盟各国规定了环丙氨嗪在畜产品中的最高残留限量为0.05 mg/kg[5]。尚未对食品中的双氰胺和缩二脲进行限量,但高剂量摄入可能对人体健康产生危害[6-7]。目前测定三聚氰胺、环丙氨嗪、双氰胺和缩二脲的方法主要有离子色谱法[8]、气相色谱-质谱法[9]、液相色谱法[10-13]、液相色谱-串联质谱[14-18]等。对于乳制品这种复杂基质样品的分析,对目标化合物的提取主要有固相萃取法[19]、液液萃取法[20-21]等,由于以上4 种化合物性质不尽相同,固相萃取法同时净化比较困难,液液萃取法净化效果一般,基质效应改善不明显,操作较复杂。分子印迹聚合物(molecularly imprinted polymers,MIPs)是对印记分子及其结构类似的客体分子具有特异选择性和识别能力的高分子功能材料,具有抗干扰性强、选择性高、稳定性好、使用寿命长等特点,适用于复杂样品的前处理[22]。近年来,磁性纳米粒子由于具有较大的表面积和独特的物理化学性质,将其与MIPs相结合制备成磁性分子印迹聚合物(magnetic molecularly imprinted polymers,MMIPs),MMIPs在完成对目标化合物的特异性识别、吸附后,只需在外加磁场的条件下即可实现与溶液的分离。其操作简单且分离时间短,使得分子印迹技术的应用领域得到进一步的发展。MMIPs作为一种新型的功能性材料,被广泛应用到细胞学、生物工程、生物医药和食品或环境中有害物质富集检测等领域[23-29]。
本实验利用磁性分子印迹技术,以缩二脲-13C2和环丙氨嗪-D4为模板分子,Fe3O4为磁性纳米颗粒,制备对三聚氰胺、环丙氨嗪、双氰胺和缩二脲具有特异性识别的MMIPs。MMIPs结合同位素稀释-超高效液相色谱-串联质谱法,实现乳及乳制品中伪蛋白的特异性分离、富集和定性及定量分析。该方法快速、灵敏、准确度高,为磁性分子印迹技术在乳制品检测中的应用提供理论依据。
1 材料与方法
1.1 材料与试剂
液态奶、酸奶、乳饮料等30 种乳制品 市购。
氯化铁、硫酸铁、氨水、油酸、甲基丙烯酸(methacrylic acid,MAA)、乙二醇二甲基丙烯酸酯(ethylene glycol dimethacrylate,EGDMA)、偶氮二异丁腈(azodiisobutyronitrile,AIBN)(均为优级醇),双氰胺标准品、缩二脲标准品、缩二脲-13C2内标(C)、三聚氰胺-三胺-15N3内标()(纯度均为99.0%) 美国Sigma-Aldrich公司;聚乙二醇6000(polyethylene glycol,PEG-6000)(分析纯) 西陇化工有限公司;聚乙烯吡咯烷酮(polyvinylpyrrolidone,PVP) 国药集团化学试剂有限公司;三聚氰胺标准品(纯度99.0%)、环丙氨嗪标准品(纯度99.0%) 美国Fluka公司;环丙氨嗪-D4(C6H6N6D4,纯度99.0%) 德国Dr. Ehrenstorfer公司。实验用水为电阻率18.2 MΩ·cm去离子水,由Milli-Q超纯水仪提供。
1.2 仪器与设备
ACQUITY UPLC超高效液相色谱仪、Xevo TQ三重四极杆串联质谱仪、Mass LynxTM色谱工作站 美国Waters公司;QGC-12T干热式氮吹仪 上海泉岛科贸有限公司;BS210S型电子天平 北京赛多利斯天平有限公司;Milli-Q超纯水仪 美国Millipore公司;MARS 6微波消解仪、聚四氟乙烯XP55T高压反应罐 美国CEM公司。
1.3 方法
1.3.1 表面修饰Fe3O4磁流体纳米颗粒的制备
利用共同沉淀法制备Fe3O4颗粒[30]。将含有FeCl3(2.0 mol/L)和FeSO4(1.0 mol/L)的混合溶液加热至50 ℃,加入氨水直到溶液pH 9,此时可见溶液中的Fe3O4深褐色沉淀。静置一段时间,移去上层清液。取出每份约8 mL的Fe3O4放入微波消解罐中,冲入氮气,拧紧旋盖放入微波消解仪,800 W、70 ℃熟化30 min。冷却到室温后,将最终的黑色沉淀进行减压抽滤,用去离子水及10%乙酸反复洗涤,去除表面的杂质。取大约2 g制备的Fe3O4颗粒,加入约40 mL去氧水,在氮气保护、80 ℃条件下加入油酸2 mL,反应30 min使Fe3O4充分被油酸包裹,降温至约50 ℃,加入PEG-6000 10.0 g,超声15 min,稀释至100 mL,冷却后即得到稳定的表面修饰Fe3O4磁流体纳米颗粒。
1.3.2 MMIPs的制备
1.0 mmol缩二脲-13C2溶于20 mL甲醇-水(1∶1,V/V)溶液中,再加入6 mmol MAA搅拌30 min,得到缩二脲-13C2与功能单体的复合物溶液;再加入20 mmol EGDMA和1.3.1节制备的1 g Fe3O4磁流体纳米颗粒,超声30 min得到预聚合复合物溶液。0.4 g PVP溶于100 mL乙醇中,溶解后充入氮气,同时加热至60 ℃。加入上述聚合复合物溶液和0.1 g AIBN,在60 ℃、氮气保护下反应24 h。冷却后在外部磁场作用下将聚合物与溶液分离,分别用去离子水、甲醇-乙酸(8∶2,V/V)溶液冲洗,直到洗脱液用液相色谱-质谱检测不到缩二脲-13C2(印迹分子)。然后将此聚合物用水冲洗,在60 ℃条件下干燥,制得可同时吸附双氰胺、缩二脲的MMIPs微球1-MMIPs[31-33]。
1.0 mmol环丙氨嗪-D4溶于20 mL甲醇-水(1∶1,V/V)溶液中,再加入8 mmol MAA,搅拌30 min,得到环丙氨嗪-D4与功能单体的复合物溶液;再按照上述步骤完成聚合反应,制得可同时吸附三聚氰胺、环丙氨嗪的MMIPs微球2-MMIPs[33]。
1.3.3 色谱条件
A C Q U I T Y U P L C B E H A m i d e色谱柱(2.1 mm×100 mm,1.7 µm);流动相A为含1%甲酸的5 mmol/L甲酸铵,流动相B为乙腈;梯度洗脱:0~2 min,3% A;2~3.2 min,3%~20% A;3.2~4.2 min,20%~30% A;4.2~4.5 min,30%~3% A;4.5~7 min,3% A。流速0.4 mL/min;柱温30 ℃;样品室温度20 ℃;进样量10 µL。
1.3.4 质谱条件
电喷雾离子源;正离子扫描;毛细管电压0.5 kV;离子源温度120 ℃;去溶剂气温度400 ℃;去溶剂气流速900 L/h;锥孔气流速50 L/h;碰撞气体为氩气,碰撞气流速0.24 mL/min;扫描方式为多反应监测模式;质谱分析参数见表1。

表1 质谱多反应监测实验条件Table1 Mass spectrometric conditions of multiple reaction monitoring (MRM)
1.3.5 混合标准溶液的配制
分别将双氰胺标准品、缩二脲标准品、三聚氰胺标准品、环丙氨嗪标准品、三聚氰胺-三胺-15N3内标物用乙腈-水(50∶50,V/V)溶液配制成1 000 µg/L的标准储备液,-18 ℃避光冷藏保存。临用时,用乙腈-水(50∶50,V/V)溶液逐级稀释双氰胺、缩二脲、三聚氰胺、环丙氨嗪标准储备溶液,配制成所需的混合标准溶液中间液,混合标准工作液以空白样品提取液稀释配制,混合标准工作液定容前需加入适量三聚氰胺-三胺-15N3内标物,使三聚氰胺-三胺-15N3内标物质量浓度为1 µg/mL。
1.3.6 样品制备过程
液态奶、酸奶、乳饮料等样品,准确称取12.5 g于三角瓶中;奶粉样品,准确称取2.0 g于三角瓶中,加入12.5 mL水溶解,加入12.5 mL乙腈,超声提取30 min。加入50 µL 1 000 µg/mL的三聚氰胺-三胺-15N3内标溶液,用乙腈-水(50∶50,V/V)溶液定容至50 mL,摇匀,滤纸过滤。称取150 mg MMIPs于烧杯中,按顺序加入4 mL甲醇,4 mL去离子水,搅拌活化。在外部磁场作用下使MMIPs与液体分离,并弃去液体。加入15 mL去离子水,3 mL样品滤液,此混合液体搅拌6 min,使目标化合物被MMIPs吸附。在外磁场作用下使MMIPs与溶液分离,弃去上层液体后,用3 mL 20%甲醇溶液清洗MMIPs表面。用甲醇-乙酸(95∶5,V/V)溶液洗脱被MMIPs吸附的目标化合物。加入1 mL甲醇-乙酸(95∶5,V/V)溶液,超声1 min,在外磁场作用下使MMIPs与溶液分离,收集液体,此操作重复6 次。6 次的洗脱液合并后约6 mL,在50 ℃氮气吹干后,加入1.0 mL流动相A溶解残渣,过0.22 µm滤膜后上机测定。
2 结果与分析
2.1 MMIPs的合成与分子识别

图1 表面修饰Fe3O4磁流体纳米颗粒在外部磁场作用下的磁分离Fig.1 Magnetic separation of surface modified Fe3O4 nanoparticles
本实验利用共沉淀法制备磁性Fe3O4纳米微球,与传统方法相比,采用微波进行熟化处理使得Fe3O4结晶程度提高;加入油酸和PEG-6000对其进行包裹,PEG-6000的疏水端和油酸的疏水端形成双层胶束,使磁性粒子能很好地分散在溶液中。添加PVP使Fe3O4纳米颗粒在水中洗涤几次或强力搅拌后仍能长时间稳定分散,并且放置4 个月以上没有沉淀产生[34]。如图1所示,在外磁场作用下伪蛋白磁性印迹聚合物迅速吸附沉降,具有很好的磁敏感性。由于三聚氰胺与环丙氨嗪有相似的化学结构,缩二脲与双氰胺也有类似化学结构,本实验选择环丙氨嗪和缩二脲的2 种同位素,即环丙氨嗪-D4和缩二脲-13C2作为模拟印迹分子合成MMIPs,可以对4 种化合物进行特异性吸附,避免了4 种化合物之间的测定干扰[35]。
制备MMIPs时,功能单体的选择、功能单体与印迹分子的配比都至关重要。碱性印迹分子常选用酸性功能单体,因此本实验在制备MMIPs时,选用酸性功能单体MAA。印迹分子与功能单体MAA混合,它们之间通过氢键作用形成功能单体-印迹分子复合物。将形成的单体-印迹分子复合物、交联剂EGDMA和表面修饰Fe3O4磁流体纳米颗粒混合,在引发剂AIBN的作用下采用热引发方式将单体-印迹分子复合物、交联剂EGDMA和Fe3O4磁流体纳米颗粒聚合,形成高聚物。最后,将三聚氰胺、环丙氨嗪、双氰胺和缩二脲印迹分子从高聚物中洗脱出来,这样高聚物上就留出与印迹分子形状、大小完全相同且具有特异识别性的孔穴。分析物中的印迹分子与高聚物上的孔穴有特异性的吸附作用,从而达到将目标物从待测物中分离的目的,见图2。

图2 MMIPs合成示意图Fig.2 Synthesis route of MMIPs
2.2 MMIPs吸附性能的测定

图3 4 种伪蛋白吸附等温曲线Fig.3 Binding isotherms of MMIPs and MNIP
通过静态吸附实验考察MMIPs对双氰胺、缩二脲、三聚氰胺、环丙氨嗪的吸附能力。制备磁性非分子印迹聚合物(magnetic non-molecular imprinted polymers,MNIP)时,除不加印迹分子外,其余步骤同1.3.2节。由图3可知,随着平衡浓度的增加,4 种伪蛋白被MMIPs吸附的含量显著增加,MMIPs对双氰胺、缩二脲、三聚氰胺、环丙氨嗪均具有较好的特异性吸附能力。MMIPs和MNIP对4 种伪蛋白的吸附量具有显著差异。这是由于MMIPs中形成了带有与印记分子互补且空间上固定排列的分子印迹位点,当印记分子进入“孔穴”中时,与其中的官能团发生对应的结合作用,能够更好地吸附目标分子。而对于MNIP来说,由于不具有特异性的印迹位点,其与底物产生结合作用属于物理吸附,无规律性,所以MNIP的吸附量变化不显著。
2.3 色谱条件与质谱条件的选择
双氰胺、缩二脲、三聚氰胺、环丙氨嗪均属于分子结构带有氨基的有机胺类强极性化合物,所以使用ACQUITY UPLC BEH Amide色谱柱,亲水相互作用色谱模式(HILIC模式)对其进行分离。电喷雾电离模式为正离子模式,故在流动相中加入甲酸加强目标化合物的质子化效果。但是实验中发现0.1%甲酸和乙腈作为流动相,进行梯度洗脱时化合物峰形和线性关系均不理想,分析原因或为流动相pH值过低,因此在流动相中引入缓冲盐,使其保持相对稳定的酸度。通过实验发现,用含1%甲酸的5 mmol/L甲酸铵溶液和乙腈对待测物进行梯度洗脱时,线性关系良好、峰形明显改善。
根据欧盟非强制执行法案2002-657-EC[36],液相色谱-串联质谱进行物质鉴定时,物质的确认需要最少4 个识别点。采用多离子反应监控模式,1 个母离子(1 个识别点),2 个子离子(每个1.5 个识别点),满足法案要求。因为4 种化合物均含碱性基团氨基,理论上在电喷雾离子源正离子模式下更易得到较强的准分子离子信号,实验中也证实了4 种化合物在电喷雾离子源正离子模式条件下[M+H]+信号最强。所以本实验采用正离子扫描,[M+H]+作为准分子离子峰(表1)。为了提高测定结果的准确性,选用三聚氰胺-三胺-15N3作为内标化合物来校正由于样品基体的干扰所带来的质谱信号的偏离,由于均属于带有氨基的有机胺类化合物,可以最大限度地减少分析误差。标准品和内标的多反应监测色谱图和的二级质谱图见图4。
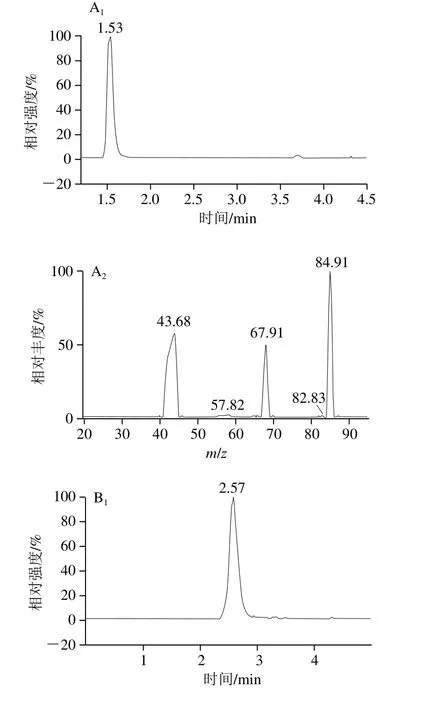
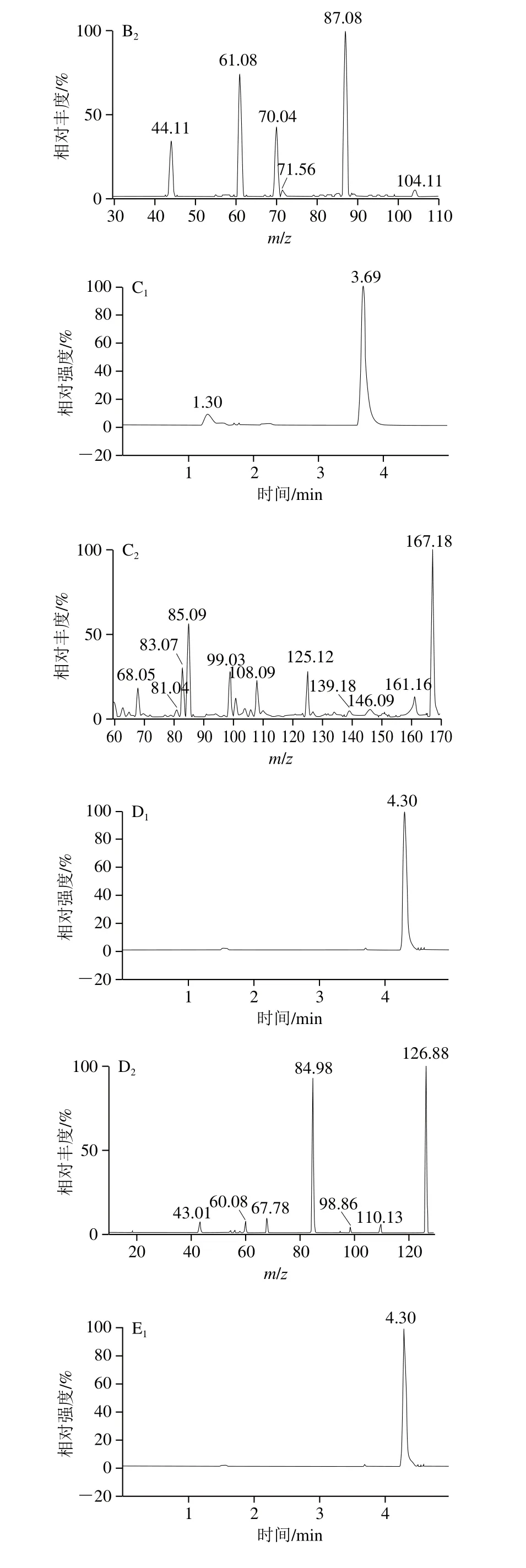

图4 多反应监测色谱图和的二级质谱图Fig.4 MRM chromatograms and tandem mass spectra
2.4 提取条件的优化
2.4.1 提取方式
双氰胺、缩二脲、三聚氰胺、环丙氨嗪均微溶于水,可溶于乙腈、甲醇等有机溶剂中,直接加入2 种MMIPs进行吸附,乳制品中大量的蛋白均会覆盖2 种MMIPs的表面,会影响2 种MMIPs对目标化合物的吸附。故选择乙腈-水(50∶50,V/V)预先除去大部分蛋白,再加入2 种MMIPs进行特异性吸附。
2.4.2 MMIPs的用量
在上述提取液(双氰胺、缩二脲、三聚氰胺、环丙氨嗪含量均为1 µg)中分别加入20、40、60、80、100、120、150、180、200 mg MMIPs,吸附完成后按照1.3.6节方法处理。结果显示,当2 种MMIPs添加量在20~150 mg时,4 种化合物的回收率均随着MMIPs添加量的增加而增加;当2 种MMIPs添加量大于150 mg时,4 种化合物的回收率不再增加。此4 种化合物的最小回收率为95.2%。由此得出添加150 mg 2 种MMIPs完全满足样品测定的需要。
2.4.3 提取时间的选择
对不同吸附时间(1~15 min)的4 种化合物的回收率进行测定。当吸附时间在1~6 min时,此4 种化合物的平均回收率从35.1%增加到95.1%,当吸附时间大于6 min时,此4 种化合物的平均回收率没有显著增加,所以确定最佳吸附时间为6 min。
2.4.4 清洗溶剂的选择
MMIPs吸附完成后首先需要清洗MMIPs表面杂质,然后将印迹孔穴中的目标化合物洗脱出来。考察不同体积分数的甲醇溶液以及乙腈溶液,分别对MMIPs表面进行清洗,并对洗脱溶剂的体积进行优化。结果表明,使用3 mL 10%甲醇溶液对MMIPs表面进行清洗,4 种化合物回收率均比较高。
2.4.5 洗脱溶剂的选择
目标化合物(印迹分子)以较弱的氢键或离子作用等非共价键与MMIPs中的特异性空穴结合。一般采用乙腈、水、甲醇-乙酸、乙腈-乙酸等相对高极性溶剂反复洗脱,即可除去印迹分子。经过比较实验,本方法加入1 mL甲醇-乙酸(95∶5,V/V)溶液对4 种化合物进行洗脱,反复6 次。为了提高提取效率,洗脱时使用超声波进行辅助提取。实验结果表明,使用该洗脱方式,4 种目标化合物均可被完全洗脱。
2.5 样品基质效应
为保证方法的通用性和适用性,本实验研究基质对分析物信号的影响,以消除干扰。在乳制品中,由于配方复杂且蛋白质等物质的存在,离子抑制十分明显。经研究采用MMIPs进行净化排除干扰物质,并结合清洗和洗脱,同时添加同位素内标抵消质谱离子化时的基质效应,从而消除了基质效应,乳粉加标样品的色谱图见图5。
图5 乳粉加标样品提取离子色谱图Fig.5 Extracted ion chromatogram of spiked milk powder sample
2.6 线性范围及检出限测定结果
按照1.3.5节方法配制一系列混合标准工作溶液,采用添加同位素内标物和空白样品提取液稀释配制标准工作液2 种方式相结合来减弱离子化时的基质效应,减小定量结果的偏差[17]。在选定的色谱条件和质谱条件下进行测定,以待测物质量浓度(X)为横坐标,以标准品与内标物峰面积比(Y)为纵坐标绘制标准溶液工作曲线。经检测发现,三聚氰胺、环丙氨嗪、双氰胺和缩二脲4 种伪蛋白各自的质量浓度与相应的峰面积比值(伪蛋白峰的面积/三聚氰胺-三胺-15N3峰面积)呈良好的线性关系,相关系数均达到0.999 0以上。
以1.3.6节方法处理添加目标化合物的空白样品,按照选定的色谱条件和质谱条件进行测定,以3 倍信噪比为方法检出限,以10 倍信噪比为方法定量限。其中检出限1与定量限1为固体样品的检出限与定量限,检出限2与定量限2为液体样品的检出限与定量限,见表2。
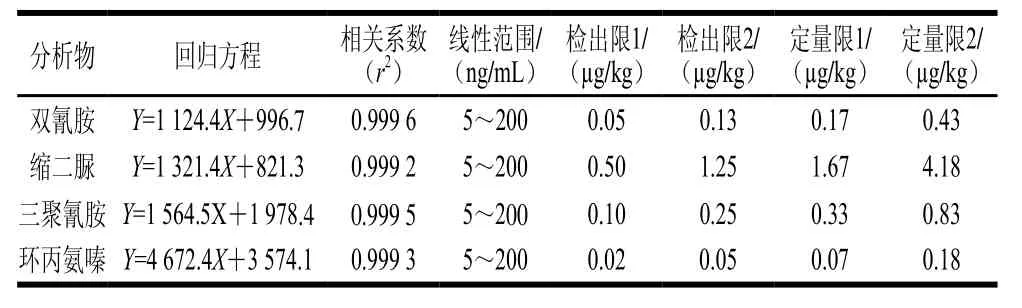
表2 方法的线性方程、线性范围、相关系数、检出限和定量限Table2 Linear equation, linear range, correlation coefficient, LOD and LOQ of the method
2.7 回收率和精密度测定结果
在不同种类的空白乳制品中添加不同水平的双氰胺、缩二脲、三聚氰胺、环丙氨嗪标准物质,其回收率、精密度结果见表3。结果显示,双氰胺的平均加标回收率为82.8%~95.5%,相对标准偏差为2.5%~9.2%;缩二脲的平均加标回收率为80.5%~94.2%,相对标准偏差为1.1%~7.2%;三聚氰胺的平均加标回收率为88.6%~95.6%,相对标准偏差为3.1%~6.9%;环丙氨嗪的平均加标回收率为83.5%~96.1%,相对标准偏差为2.1%~7.5%。
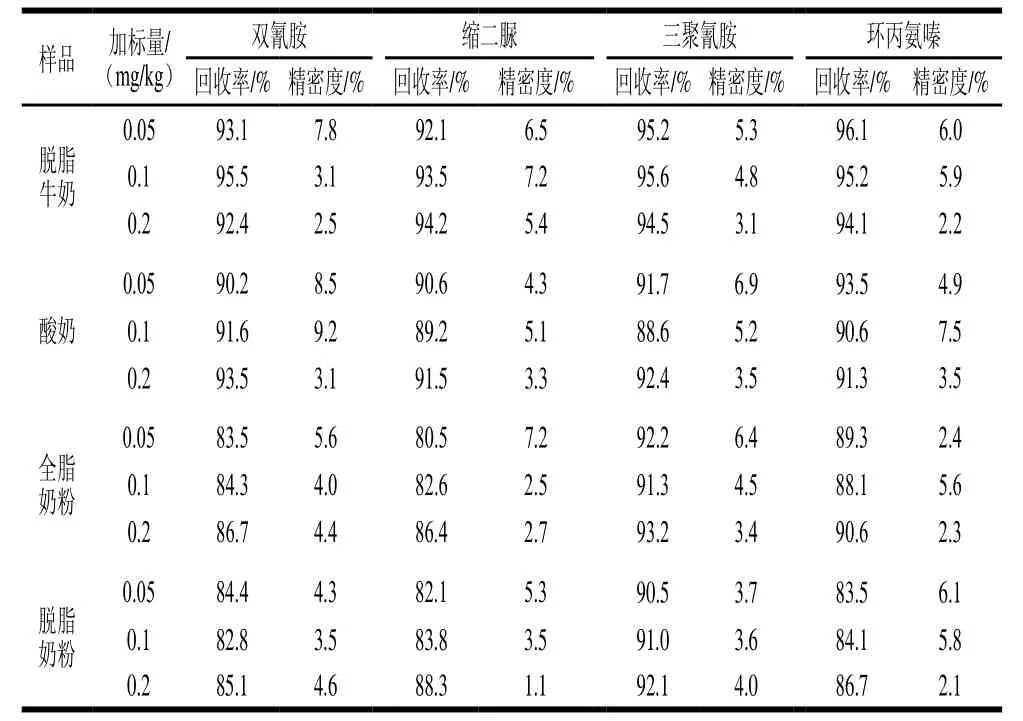
表3 不同乳制品中回收率与精密度实验结果(n=6)Table3 Recovery and precision for 6 replicate determinations of four pseudo proteins in different samples (n= 6)
2.8 实际样品测定结果
对市售的30 种乳制品进行分析测定,利用本方法对每个样品重复测定3 次,结果表明三聚氰胺、环丙氨嗪、双氰胺和缩二脲含量均小于方法检出限。
3 结 论
本实验以缩二脲-13C2和环丙氨嗪-D4为印记分子,MAA为功能单体,EGDMA为交联剂,Fe3O4为磁性组分成功制备出对三聚氰胺、环丙氨嗪、双氰胺和缩二脲有特异选择吸附性能MMIPs。此外,该聚合物具有很好的磁性,在完成对目标物的吸附后,可以在外加磁场作用下实现动态快速分离,使提取过程更加方便、快捷。应用超高效液相色谱-串联质谱联用技术,对提取出来的三聚氰胺、环丙氨嗪、双氰胺和缩二脲进行色谱分离、定性、定量分析,其结果准确、可靠。
猜你喜欢
中国化肥信息(2022年2期)2023-01-02 12:17:29
中国化肥信息(2022年8期)2022-11-30 06:20:14
陶瓷研究(2022年3期)2022-08-19 07:15:18
云南画报(2021年10期)2021-11-24 01:06:56
磷肥与复肥(2020年6期)2020-07-16 10:46:08
小学生优秀作文(高年级)(2018年4期)2018-09-11 01:23:22
中国化肥信息(2018年2期)2018-08-23 09:09:16
粘接(2016年10期)2016-11-10 05:55:24
中国化肥信息(2016年41期)2016-05-17 04:25:58
现代纺织技术(2015年2期)2015-02-28 14:03:11
