
HPLC测定毛蕊花糖苷原料药含量的不确定度评估△
2019-01-16 00:59毛乐静霍仕霞孔征谭雪闫明
中国现代中药 2018年12期
毛乐静,霍仕霞,孔征,谭雪,闫明
(1.新疆医科大学 药学院,新疆 乌鲁木齐 830011;2.新疆维吾尔自治区维吾尔医药研究所,新疆 乌鲁木齐 830049)
毛蕊花糖苷为苯乙醇苷类化合物,具有抗衰老、增强免疫力、抗老年痴呆等作用[1-2]。笔者课题组已从新疆特色资源管花肉苁蓉中提取分离纯化制备得到了高纯度的毛蕊花糖苷原料药,建立可靠的质控方法是保证该原料药质量的关键,笔者依据2015年版《中华人民共和国药典》一部肉苁蓉含量测定项下方法,进行了毛蕊花糖苷原料的含量测定,在测定的过程中,发现该方法存在很多不确定的因素,在称量、溶液稀释、测定及方法学考察等每一个环节都存在不确定的因素,这些不确定的因素造成最终的测量值都会存在不能肯定的部分,称为不确定度[3]。不确定度与随机性、模糊性、不完全性有关。测量的不确定度在分析化学领域是一个非常重要的概念,是评价实验结果可信赖程度的指标。为了增加实验结果的可比性,需要将其更加广泛地应用到分析实验中[4]。作者针对毛蕊花糖苷原料药含量测定每个环节存在的不确定因素进行分析,并对每个环节所产生的不确定度加以归纳,从中分析各个环节的不确定度对最终测定结果的影响,评估该含量测定方法的可靠可信度,使含量测定结果更有意义,为毛蕊花糖苷原料药质量控制和客观评价提供科学依据。
1 仪器与材料
1.1 仪器
岛津高效液相色谱仪:LC-20 AB泵,SPD-M 20 A二极管阵列检测器,CBM-20 A系统控制器,CTO-20 A柱温箱,SIL-20 A自动进样器,DGU-20 A3在线脱气装置;SQP-224-1CN天平(赛多利斯科学仪器有限公司);SK 8210 HP型超声清洗器(上海冠特超声仪器有限公司);SZ-93自动双重纯水蒸馏器(上海亚荣生化仪器厂);BDS Hypersil C18(250 mm×4.6 mm,5 μm)色谱柱。
1.2 材料
毛蕊花糖苷对照品(中国食品药品检定研究院,批号:111530-201208,纯度:92.5%);甲醇:HPLC级,Sigma-Aldrich公司;水为超纯水;甲酸(天津市富宇精细化工有限公司,批号:20140818)。
毛蕊花糖苷原料药(新疆维吾尔自治区维吾尔医药研究所,批号:20130228、20130311、20130323、20130405、20130417、20130426、20130511、20130520、20130526、20130611)。
2 方法
2.1 色谱条件及系统适用性
BDS Hypersil C18(250 mm×4.6 mm,5 μm)色谱柱;检测波长:330 nm[5];流动相:甲醇-0.2%甲酸水溶液(35∶65)[6];流速:1.0 mL·min-1;柱温:30 ℃;在上述条件下进样分析,毛蕊花糖苷与其他组分实现基线分离,分离度大于1.5,理论板数大于3000。
2.2 对照品溶液的制备
取毛蕊花糖苷对照品20 mg,精密称定,置25 mL量瓶中,加流动相[甲醇-0.2%甲酸溶液(35∶65)]稀释至刻度,摇匀,制成每1 mL含0.8 mg毛蕊花糖苷的对照品储备液。精密吸取对照品储备液1 mL置10 mL量瓶中,加流动相溶解定容,摇匀,滤过,取续滤液,即得。
2.3 供试品溶液的制备
取毛蕊花糖苷原料药20 mg,精密称定,置100 mL量瓶中,加流动相溶解稀释至刻度,摇匀,精密吸取5 mL至10 mL量瓶中,加流动相溶解定容,摇匀,滤过,取续滤液,即得。
2.4 线性关系考察
精密吸取上述对照品溶液适量,分别制成毛蕊花糖苷质量浓度为5、10、20、40、60、80、120 μg·mL-1的系列对照品溶液,按2.1项下方法测定,分别以对照品溶液浓度(C,μg·mL-1)为横坐标(X),峰面积积分值为纵坐标(Y),绘制标准曲线并进行回归处理,得毛蕊花糖苷标准曲线回归方程:Y=207 67X-1 329.5,r=0.999 6,线性范围为5~120 μg·mL-1,结果表明在5~120 μg·mL-1毛蕊花糖苷峰面积与质量浓度具有良好的线性关系。
2.5 精密度考察
取毛蕊花糖苷原料药供试品(批号:20130228)6份,按供试品项下的方法制备供试品溶液,照含量测定色谱条件下进样,记录峰面积。结果RSD为1.3%(n=6)。
2.6 稳定性考察
取同一批毛蕊花糖苷原料药样品(批号:20130228),处理条件分别为室温和4 ℃冷藏,分别于0、1、2、4、6、8、12、24、36、48、72、96 h进行含量测定,结果室温放置条件下RSD为1.2%(n=12),冷藏条件下RSD为1.4%(n=12),表明供试品溶液在96 h内稳定性良好。
2.7 检测限及定量限测定
配制质量浓度适当的毛蕊花糖苷对照品溶液,进样10 μL,当信噪比S/N为3时,其检测质量浓度约为0.120 0 μg·mL-1;当信噪比S/N为10时,其定量限为0.375 0 μg·mL-1。
2.8 加样回收率试验
取毛蕊花糖苷原料药同一批样品(批号:20130228),配制低、中、高3个水平的溶液,平行3份。按2.1项下色谱条件进样测定,记录峰面积。结果低、中、高质量浓度的供试品溶液加样回收率分别为101.1%、98.2%、99.5%(n=3),平均加样回收率为99.6%(n=9),RSD为1.8%。
2.9 样品含量测定
制备10批样品,按供试品溶液制备法处理,按2.1色谱条件下测定,利用标准曲线计算含量,得样品平均质量分数为99.0%。
3 不确定度分析
用GUM法评定测量不确定度的一般流程:分析不确定度来源和建立测量模型→评定标准不确定度→计算合成标准不确定度→确定扩展不确定度→报告测量结果[7]。
3.1 不确定度来源
高效液相色谱法测定分析过程中的不确定度主要来源于几个方面:称量过程、稀释过程、仪器、对照品纯度、回收率、标准曲线以及其他因素[8-9]。如图1所示。

图1 不确定度来源分析
3.2 数学模型的建立
采用标准曲线对毛蕊花糖苷原料药进行含量测定,毛蕊花糖苷含量计算公式如下[10]:
(1)
式中:X为样品中毛蕊花糖苷含量(mg·g-1);C样为根据标准曲线求算的样品质量浓度(mg·L-1);P对为对照品的纯度(%);V样为样品稀释体积(mL);W样为供试品的取样量(g);D为对照品溶液的稀释倍数;R为方法回收率。
3.3 各不确定度的分析和评定

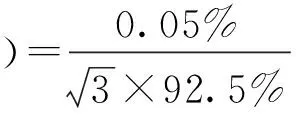
(2)
式中:p对为对照品纯度。
3.3.2 称量引入的不确定度(B类不确定度) 对照品与样品称量引入的不确定度[12]:称量使用SQP-224-1 CN天平(称量范围220 g,精度0.1 mg),称样量分别为20 mg。
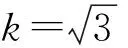
(3)
(4)
(5)
式中:u1为天平示值的不确定度;u2为天平称量重复性引入的不确定度;u为天平引入的不确定度;urel(w)为天平称量引入的相对标准不确定度;m为质量。
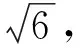
吸量管的不确定度:配制对照品溶液需使用1.0、2.0、5.0 mL吸量管,20 ℃时1.0、2.0、5.0 mL A级吸量管的允差分别为±0.008、±0.012、±0.025 mL[14]。配制对照品溶液由吸量管引入的不确定度按公式(7)计算得为3.278×10-3。
温度的不确定度:容量器皿校准温度为20 ℃,实验室温度为25 ℃,(20±5)℃时水的体积膨胀系数为2.080×10-4。故对照溶液配制过程中由温度引入的不确定度按公式(8)计算得为6.467×10-5。对照品溶液稀释过程引入的相对不确定度按公式(9)计算得为3.316×10-3。

(6)

(7)
(8)

(9)
式中:urel(V对)为容量瓶体积引入的不确定度;n为稀释次数;u10为10 mL容量瓶引入的不确定度;u25为25 mL容量瓶引入的不确定度;urel(Vp)为吸量管引入的不确定度;urel(VT)为温度对容量瓶影响引入的不确定度;u(T25)为温度对25 mL容量瓶影响引入的不确定度;u(T10)为温度对10 mL容量瓶影响引入的不确定度;u对为对照品溶液稀释引入的不确定度。
3.3.4 样品稀释过程引入的不确定度(B类不确定度) 容量瓶体积的不确定度[15]:20 ℃时100 mL容量瓶的容量允差为±0.1 mL。由容量瓶引入的相对不确定度为4.082×10-4。由温度引入的不确定度为6.004×10-6。由温度引入的不确定度可忽略不计,样品稀释过程引入的相对不确定度为4.082×10-4。
3.3.5 与标准曲线有关的不确定度(A类不确定度) 采用表1数据按公式(10)和公式(11)得出Y=20 767X-1 329.5(Y表示峰面积,X表示质量浓度),r=0.999 6,则峰面积测量的标准残差s为11 808.99,n=7[16]。
应用该曲线测量时,重复3次测量样品处理液,即p=3,测得样品平均质量分数为99.0%。

表1 毛蕊花糖苷对照品溶液的HPLC峰面积
(10)
sxx=∑(cj-ca)2=24 245.618 62
(11)
(12)
(13)
式中:s为标准曲线的标准残差,b1为斜率,p为样品的测定次数,n为标准曲线的浓度点个数,c0为标准曲线计算的样品浓度,ca为制作标准曲线的算术平均浓度,cj为校正曲线的单点浓度;c为标准溶液浓度均值;sxx为残差平方和;yi为峰面积;xi为浓度。
由公式(12)和公式(13)计算得到的不确定度和相对不确定度为0.402 9和9.101×10-3。
3.3.6 测量重复性引入的不确定度(A类不确定度)
3.3.6.1 对照品溶液色谱峰面积的相对不确定度 取20 μg·mL-1的对照品溶液,连续进样6次[17],记录峰面积分别为379 605、379 402、379 682、369 503、379 554、379 632。
(14)
(15)

(16)
平均峰面积A为377 896,RSD=1.1%。标准偏差按公式(15)计算得为4041,实验标准偏差按公式(14)计算得为1649。对照品溶液相对不确定度按公式(16)计算得为4.446×10-3。
3.3.6.2 样品溶液色谱峰面积的相对不确定度 制备6份供试品溶液,进样测定,峰面积分别为410 489、413 273、410 655、411 122、413 165、399 500,平均峰面积为409 700,RSD为1.3%。标准偏差为366 835,实验标准偏差为149 759,按公式(17)计算引入的不确定度为5.124×10-4。
(17)
3.3.7 回收率的不确定度(A类不确定度) 分别求算高、中、低质量浓度的不确定度[18]。高质量浓度的加样回收率分别为100.0%、99.1%、99.5%,中浓度的加样回收率分别为97.6%、99.0%、98.0%,低浓度的加样回收率分别为102.8%、102.2%、98.4%。高、中、低质量浓度的RSD为0.468 0%、0.709 5%、0.235 0%,高质量浓度:标准偏差为0.465 7,实验标准偏差为0.268 9,引入的不确定度为1.911×10-3。中质量浓度:标准偏差为0.696 6,实验标准偏差为0.402 2,引入的不确定度为2.897×10-3。低质量浓度:标准偏差为2.376,实验标准偏差为1.372,引入的不确定度为9.594×10-4。
3.3.8 HPLC引入的相对不确定度(B类不确定度) 岛津高效液相色谱仪检定证书给出定量测量重复性RSD为0.3%(n=6)[19],按公式(18)计算得相对不确定为1.225×10-3。
(18)
3.3.9 合成相对标准不确定度(U) 将上述不确定度分量依据含量计算公式,对不确定度合成,按公式(19)计算得不确定度为0.046 8,取包含因子k=2,此时对应的置信概率为95%,毛蕊花糖苷原料药质量分数为99.0%,质量分数的标准不确定度为4.682,其扩展不确定度为9.364。
(19)
3.4 不确定度分量
该方法引入的总体不确定度为0.046 8,各不确定度分量所占比重如表2所示,比重越高,说明该因素引入的不确定度越大,在实验操作过程中需要严格控制。

表2 HPLC测定毛蕊花糖苷原料药含量的不确定度评价
4 讨论
4.1 实验过程中各不确定的分类
不确定度指由于测量误差的存在,对被测量值的不能肯定的程度,也表明测定结果的可信赖程度。不确定度越小,所述结果与被测量的真值愈接近,质量越高,水平越高,其使用价值越高;不确定度越大,测量结果的质量越低,水平越低,其使用价值也越低[20-21]。在ISO指南中将这些不同种类的分量分别划分为A类评定和B类评定。本文中A类不确定度包括标准曲线的不确定度、测量重复性的不确定度、加样回收率的不确定度;B类不确定度包括对照品纯度的不确定度、称量引入的不确定度、稀释过程引入的不确定度、高效液相引入的不确定度。评估不确定度时,我们需要密切注意产生不确定度的所有可能来源,然后集中精力分析最大的不确定度分量。
4.2 降低不确定度的方法
本研究在对毛蕊花糖苷原料药进行含量测定的过程中,分析评价了从对照品配制到分析测定各个环节的不确定度,并分析了每个环节不确定度所占的比重,其中称量造成的不确定度比重最大,占到70%,取样量的大小及所使用的分析天平的精度直接决定了不确定度的大小,分析时尽量在允许范围内,增大称样量,选择符合要求的容器;第二是含量测定过程中,标准曲线所产生的不确定度,标准溶液的浓度、标准曲线的斜率、测定的峰面积不确定度,对标准曲线有一定的影响[22];第三对照品和供试品测定过程中,色谱峰峰面积测定[23]、重复性及重现性考察等都会产生不确定度,这就要求分析人员在操作过程中,尽可能减少操作所造成的不确定;此外对照品的纯度、稀释过程、仪器允差[24]也会引入一定的不确定度,如配制溶液用到的移液管、容量瓶、稀释的倍数、对照品的选择,这些都是可以人为控制的因素。仪器选择上,尽量选择允差较小的设备。本研究的结果分析了毛蕊花糖苷原料药含量测定过程中存在不确定度,旨在对分析中的不确定度有一个定性和定量的认识,为后期优化毛蕊花糖苷原料药的质控方法提供科学的依据。
猜你喜欢
中西医结合心脑血管病杂志(2022年20期)2022-11-08
中国中医药现代远程教育(2022年17期)2022-09-26
中国马铃薯(2022年2期)2022-07-05
中草药(2022年11期)2022-05-31
中草药(2022年9期)2022-05-06
科学技术创新(2021年19期)2021-07-16
中成药(2020年8期)2020-09-15
科技创新与应用(2017年1期)2017-05-11
科技与创新(2016年10期)2016-05-28
吉林农业·下半月(2014年12期)2014-12-25
