
片状纳米结构CeO2晶体的制备及其吸附CO2性能
2019-01-14 06:00赵博生史会虎张建斌
无机化学学报 2019年1期
赵博生 史会虎 马 良 杜 宏 张建斌*,
(1内蒙古工业大学化工学院,呼和浩特 010051)
(2内蒙古自治区CO2捕集与资源化工程研究中心,呼和浩特 010051)
In the past years,steel plants,thermal power plants and chemical plants emitted large amounts of CO2gas every year,causing environmental pollution to become more serious,which had become the focus of global attention[1-3].In general,amine solutions were often used to treat CO2in industrial exhaust gases[4].However,the amine solutions presented high volatile and corrosive,which increased the treatment cost of industrial exhaust gas[5].To solve the shortcomings of the amine solutions,more stable and economical method for CO2capture should be developed.A new type of solid adsorbent had stable structure,chemical properties and efficient cycle performance,which could be used to capture CO2in industrial exhaust,such as metal oxides[6-10],carbon-materials[11-13],silicamaterials[14-17], metal organic frameworks[18-20]and zeolites[21-23].The main reason for studying metal oxides was that metal oxides had a higher true density than other materials[24].Especially,CeO2materials could be used as the good solid CO2adsorbent due to its strong Lewis base sites[25-26].Normally,CeO2crystals were prepared by calcining CCPs,which had been synthesized through many methods,such as hydrothermal methods[27-28], sol-gel methods[29-30], precipitation methods[31],solvothermal methods[32-33]and microwave methods[34-35].However,these synthetic methods had a long reaction time and required additional template additive,resulting in high cost[36-38].
Based on the above discussion,this work proposed a simple synthesis method without additional additives for facile preparation of nano-structure flakelike CCPs,which mixed CeCl3solution with CO2SM at room temperature and stirred at a certain rotational speed.The nano-structure flake-like CeO2crystals were obtained by calcining CCPs for 4 h at 500℃.In the synthesis process,the CO2SM played a significant role,which provide CO32-and act as dispersant and structure-directing agent.The effect of three factors on the morphology and size of flake-like CCPs were studied,including CO2SM dosage,Ce3+concentration and stirring time.The flake-like CeO2crystals not only had good reduction performance,but also could capture CO2and reached 0.554 mmol·g-1at room temperature.Fig.1 shows the entire preparation process.CCPs prepared through stirring CO2SM and Ce3+and CeO2was obtained by calcining CCPs,which could adsorb CO2.CO2SMwassynthesized by absorbing CO2with ethylene glycol (EG)and 1,2-ethanediamine(EDA).

Fig.1 Entire preparation process
1 Experimental
1.1 Materials
EG and EDA was analytical grade.99.999%(V/V)compressed CO2was obtained from the Standard Things Center.Cerium バ chloride heptahydrate(CeCl3·7H2O)was purchased from Aladdin Company.The CO2SM was synthesized by the EDA+EG system uptaking CO2[39].
1.2 Preparation of CCPs and CeO2 crystals
At room temperature,0.1~0.5 g CO2SM and 50 mL 0.01~0.1 mol·L-1Ce3+solution were mixed on the magnetic stirrer and stirred at 1 000 r·min-1for 0.5~3 h to prepare CCPs.After the reaction,the CCPs were washed using double-distilled water and ethanol while being vacuum filtered and the precipitate was collected after repeated washing three times.To obtain the dried CCPs,the filtered precipitate was dried at 120℃for 2 h.Finally,the nano-structure flake-like CeO2crystals were prepared by calcining CCPs at 500℃ with the heating step of 5℃·min-1for 4 h in air.
1.3 Characterization
The CCPs and CeO2crystals were investigated by a Quanta FEG 650 scanning electron microscopy(SEM)with an accelerating voltage of 20 kV and a JEM-2100 high magnification transmission electron microscopy (HR-TEM)with an accelerating voltage of 200 kV.Their X-ray diffraction (XRD)patterns were collected on a Siemens D/max-RB powder X-ray diffract mete with Cu Kα radiation (λ=0.154 059 8 nm)over the 2θrange of 10°~80°with the scanning rate of 0.05°·s-1operated at 40 kV and 40 mA.Fourier transform infrared spectroscopy (FT-IR)was recorded on a Nexus 670 infrared spectrophotometer.Isotherms were analyzed using 3H-2000PS2 BET instrument with the Barrett-Joyner-Halenda (BJH)theory to give the pore parameters,including Brunauer-Emmett-Teller (BET)surface area,total pore volume and average pore size.Entzsch-Sta 449 thermogravimetry analysis (TGA)was employed to measure the weight percentage of CCPs.
1.4 H 2 temperature-programmed reduction(H 2-TPR)
The H2-TPR of CeO2crystals was measured through the TP-5080 equipment possessed a thermal conductivity detector (TCD).First,50 mg CeO2crystals were placed in a quartz tube of the reactive furnace for heat treatment.The reactive furnace was heated to 400℃through the temperature-programmed method for 30 min using N2as a shielding gas(30 mL·min-1)to remove physically adsorbed H2O and/or CO2,and then cooled to room temperature.Then,hydrogen and nitrogen were mixed into a ratio of 5%(V/V)H2,raisingthetemperature of the reactive furnace from room temperature to 1 000℃ with 10℃·min-1to reduce CeO2crystals.Finally,the soft file of TP-5080 equipment was used to calculate the consumed amount of H2by CeO2crystals.
1.5 CO2 temperature-programmed desorption(CO2-TPD)
The alkalinity of CeO2crystals was measured by CO2-TPD through the TP-5080 equipment possessed a TCD.First,100 mg of CeO2crystals were placed in a quartz tube of a reaction furnace and heated at 400℃for 30 min to perform heat treatment.Then,the pretreated CeO2crystals was adsorbed with pure CO2gas at room temperature,120 and 700℃for 30 min.After the adsorption,He gas was used to purge the CO2in the pipeline.The reaction furnace heated from room temperature to 1 000℃ with 10℃·min-1.Finally,the soft file of TP-5080 equipment was used to calculate the amount of CO2consumed by CeO2crystals.

Fig.2 SEM images of CCPs under different CO2SM dosages:(A)0.1,(B)0.2,(C)0.3,(D)0.4 and (E)0.5 g
2 Results and discussion
2.1 Preparation of CCPs
In the experimental process,the influence of three factors of CO2SM dosage,stirring time and Ce3+ion concentration on the morphology and size of CCPs were systemically investigated.
2.1.1 CO2SM dosages
To study the effect of CO2SM dosage on the morphology and size of CCPs,the different amount of CO2SM (0.1 ~0.5 g)was mixed with 50 mL water solution of 0.03 mol·L-1Ce3+using the magnetic stirrer.After stirring for 1 h at 1 000 r·min-1,the CCPs were finally obtained.The size of CCPs were measured from SEM images.
According to the SEM images of CCPs in Fig.2,it could clearly observe the nano-structure flake-like CCPs.Through measuring the length and width of CCPs,it could be concluded that the size of the flake-like CCPs gradually increased from 5.08μm×0.96μm in Fig.2A to 5.57 μm×1.46 μm in Fig.2D when the amount of CO2SM increased from 0.1 to 0.4 g.When CO2SM dosage continued to increase by 0.5 g,the size of flake-like CCPs reduced by 5.35 μm×1.29 μm,which was due to the formation of CCPs′fracture.The 0.1 g CO2SM could be used to prepare the minimum size of 5.08 μm×0.96 μm flake-like CCPs with the homogeneous morphology.Therefore,0.1 g CO2SM was selected for next experiment.Analysis of the effects of CO2SM dosages on the morphology and size of CCPs,the more CO2SMwas dissolved in the solution,the more CO32-would be generated and high nucleation rate of crystals.Therefore,as the CO2SM dosages increased,the average diameter could gradually increase.However,when the CO2SM dosages was too much,the morphology of CCPs would be broken to make the size smaller.
2.1.2 Ce3+concentration
To study the effect of Ce3+concentration on the morphology and size of CCPs,the 50 mL different concentrations of Ce3+aqueous solution (0.01~0.1 mol·L-1)was mixed with 0.1 g CO2SM using the magnetic stirrer.After stirring for 1 h at 1 000 r·min-1,the CCPs were finally obtained.The size of CCPs were measured from SEM images.
According to the SEM images of CCPs in Fig.3,it could be clearly observed that the Ce3+concentrations did not affect the morphology of CCPs.When the Ce3+concentration was 0.01 mol·L-1,it could be seen that some small fragment CCPs were not uniform enough and the reunion phenomenon was serious.When the Ce3+concentration increased from0.03~0.1 mol·L-1,the size of flake-like CCPs increased gradually from 5.08 μm×0.96 μm in Fig.3B to 5.89μm×1.22 μm in Fig.3E.At the Ce3+concentration of 0.03 mol·L-1,the minimumsize of flake-like CCPs with the homogeneous morphology could be obtained.Therefore,0.03 mol·L-1Ce3+aqueous solution was used for next experiment.Analysis of the effects of Ce3+concentration on the morphology and size of CCPs,the higher Ce3+concentration,the higher the content of Ce3+would generate more CCPs aggregates.Therefore,the size of the formed CCPs would become larger.Ce3+concentration did not change the morphology of CCPs.

Fig.3 SEM images of CCPs under 50 mL different concentrations of Ce3+aqueous solution:(A)0.01,(B)0.03,(C)0.05,(D)0.08 and (E)0.1 mol·L-1
2.1.3 Stirring time
To study the effect of stirring time on the morphology and size of CCPs,the 50 mL 0.03 mol·L-1Ce3+aqueous solution was mixed with 0.1 g CO2SM at different stirring times (0.5~3 h)using the magnetic stirrer.After stirring at 1 000 r·min-1,the CCPs were finally obtained.The size of the CCPs were measured from SEM images.
According to the SEM images of CCPs in Fig.4,it could be clearly observed that the stirring time also did not affect the morphology of flake-like CCPs.By measuring the size of the flake-like CCPs prepared at different stirring time,it could be concluded that the size of the flake-like CCPs increased gradually from the value of 5.08 μm×0.96 μm in Fig.4A to 7.19 μm×1.48 μm in Fig.4E with the increase of stirring time.The minimum size of the flake-like CCPs could be prepared at 0.5 h.Therefore,0.5 h was chosen as the optimum stirring time.Analysis of the effects of stirring time on the morphology and size of CCPs,as time increased,the resulting CCPs aggregates also increased.Therefore,the size of the formed CCPs would become larger.Stirring time did not change the morphology of CCPs.
According to the XRD patterns of the CCPs in Fig.5(a),it could be observed that almost no diffraction peaks appeared in XRD patterns,indicating the prepared CCPs were amorphous.According to FT-IR spectrum of CCPs in Fig.5(b),it could be observed that the FTIR peak at 3 430 cm-1occurred due to the O-H stretching vibration of-OH group and the peak shape had not changed with the time increasing[40-41].The FT-IR absorption peak at 1 490 cm-1was due to the double asymmetrical tensile vibration of the CO32-group and the peak shape changed from package peak to sharp peak with the time increasing[42-43].The other FT-IR peaks at 691,754 and 843 cm-1were due to bending vibration of the CO32-group[44-45].Analysis of FT-IR spectra showed that CCPs contained-OH and CO32-groups.

Fig.4 SEM images of CCPs under different stirring times:(A)0.5,(B)0.75,(C)1,(D)2 and (E)3 h
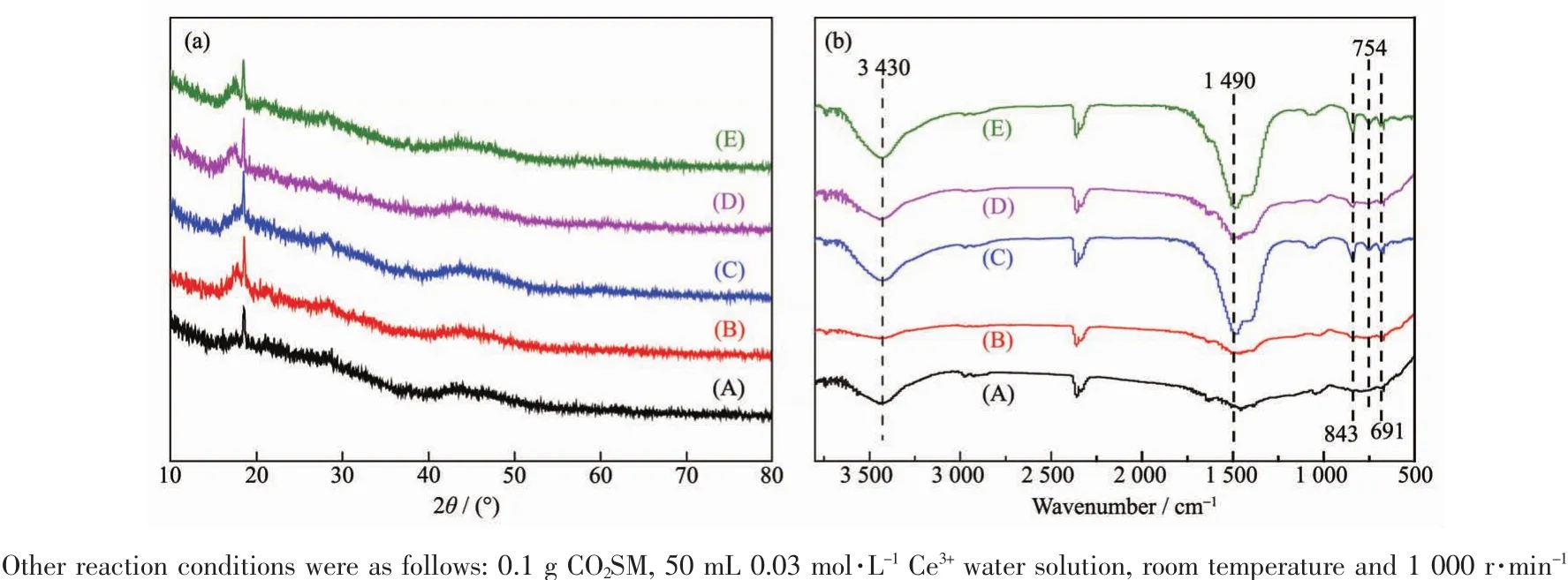
Fig.5 (a)XRD patterns and (b)FT-IR spectra of CCPs obtained after different stirring times:(A)0.5,(B)0.75,(C)1,(D)2 and (E)3 h
Through the above three factors,the optimum preparation conditions were confirmed at 0.1 g CO2SM with 50 mL 0.03 mol·L-1Ce3+aqueous solution at 1 000 r·min-1for 0.5 h at room temperature.
2.2 Possible formation mechanism of CCPs
Fig.6 showed the possible formation mechanism of nano-structure flake-like CCPs,in which the CO2SM played a crucial role in crystal nucleation and growth.The CO2SM not only provided a source of CO32-but also decomposed EG+EDA in aqueous solution[46].Ce3+was surrounded by EG/EDA because of the strong electrostatic interaction between EDA and EG[47].This hindered the collision between Ce3+and CO32-,and eventually formed small CCPs aggregates.Small CCPs aggregates accumulated into the flake-like subunit structure due to hydrogen bonding between EDA and EG molecules.Moreover,the CO2SM could also act as a dispersant and structure-directing agent[48].Finally,the flake-like subunit structure could be freely assembled into flake-like CCPs.

Fig.6 Possible formation mechanism of CCPs
2.3 TG-DTG
According to the typical TG-DTG curves of asprepared CCPs in Fig.7,it could be found two distinct weight loss stages and the decomposition process of CCPs.The weight loss of the first stage occurred in the range of 50~314 ℃ and the weight loss of 5.51%was due to the evaporation of water and/or organic compounds that were physically or chemically absorbed on the surface of CCPs.The weight loss of the second stage occurred in the range of 314~684 ℃and the large weight loss of 23.54%was due to the thermal decomposition of CCPs released CO2.According to the DTG curve,it could be observed that the thermal decomposition of CCPs to CeO2was the endothermic process and the maximum endothermic peak occurred at 509℃.According to the TG-DTG curves,CeO2crystals were prepared by calcining CCPs at 500℃for 4 h.
2.4 XRD,SEM and TEM
XRD patterns of CeO2crystals were performed to determine the crystal form.According to the XRD patterns,the diffraction peaks at 2θ=28.560°,33.160°,47.550°,56.480°,59.291°,69.510°and 76.780°corresponded to (111),(200),(220),(311),(222),(400)and (331)planes of face-centered cubic (FCC)CeO2crystals (PDF No.43-1002)[49].
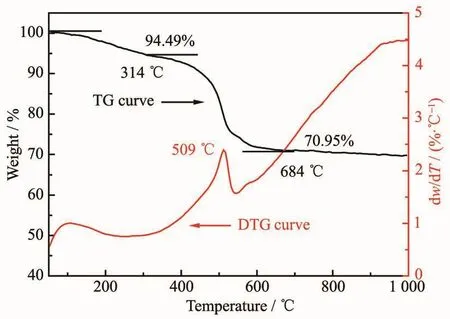
Fig.7 TG-DTGcurves of as-prepared CCPs

Fig.8 XRD patterns of flake-like CeO2 crystals
Fig.9 was the SEM image of flake-like CeO2crystals.By comparing SEM images before and after calcining,it could be concluded that the calcination of CCPs at 500℃ did not change the original morphology but only decreased in size from 5.08μm×0.96 μm to 4.94 μm ×0.92 μm.The thickness of flake-like CeO2crystals were measured at 0.06μm.
From Fig.10A,the size of CCPs was 4.66μm×0.86μm.The lattice spacing of CCPs were not clear and the crystal planes were complicated.The selected area electron diffraction (SAED)patterns of CCPs were composed of several bright rings,indicated that CCPs were amorphous.From Fig.10B,there were two flake-like CeO2crystals with dimensions of 4.93μm×0.64 and 4.91 μm×0.67 μm.The flake-like CeO2crystals prepared after calcining became more transparent and the lattice spacing was clear.By measuring the distance between the black stripes,it could be concluded that the lattice spacing of 0.31 and 0.19 nm were equivalent to the (111)and (220)planes of the face-centered cubic CeO2crystals,respectively[50].According to the SAED patterns,the distance from the bright spot to the center was measured to be 0.313 and 0.190 nm corresponding to the (111)and (220)planes of CeO2crystals,respectively.Therefore,it was shown that the flake-like CeO2crystals had a typical poly-crystalline structure.

Fig.9 SEM image of flake-like CeO2 crystals

Fig.10 TEM images of CCPs (A)and CeO2 crystals (B)
2.5 N2 adsorption-desorption isotherm
From Fig.11,it could be observed that the relative pressure P/P0between 0.4 and 1.0 appeared a hysteresis loop.According to the IUPACclassification,the isotherm of the CeO2crystals was attributed to the typical typeⅣisotherm[51].Therefore,it could conclusive that CeO2crystals possessed mesoporous structure.The BETsurface area,total pore volume and BJH pore diameter of the CeO2crystals were measured as 9 m2·g-1,0.02 cm3·g-1and 11.6 nm,respectively.
2.6 H 2-TPR
From Fig.12,it could be observed that the function of the H2consumption of CeO2crystals recorded in the range from 50~900 ℃.Two strong absorption peaks occurred at 516.7 and 771.9℃.According to the software corresponding to the TP5080 instrument,the amount of H2consumption of CeO2crystals could be calculated as 6.362 mmol·g-1,indicating that the CeO2crystals had good reduction performance[52].
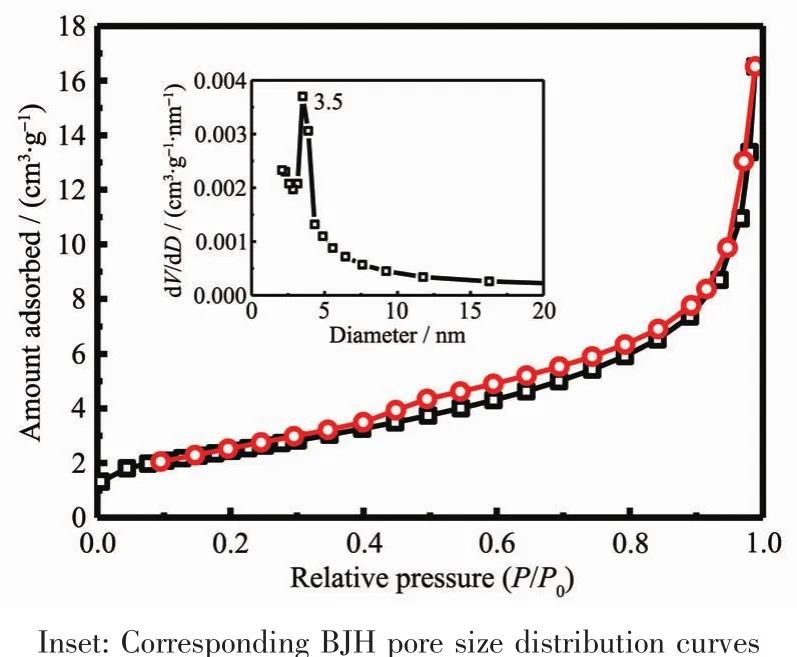
Fig.11 N2 adsorption-desorption isotherms of as-obtained flaky-like CeO2 crystals

Fig.12 H2-TPRprofile of as-prepared flake-like CeO2 crystals
2.7 CO2-TPD
From Fig.13,it could be observed that CO2consumption of CeO2crystals is a function of temperature in the range of 50~900 ℃.According to the software corresponding to the TP5080 instrument,the amount of CO2consumption of the CeO2crystals at different temperatures(room temperature,120 and 700℃)could be calculated as 0.554,0.527 and 0.059 mmol·g-1,respectively.Adsorption capacity at room temperature was higher than previous reports(7.8,11.6 mg·g-1)[52-53].According to the analysis of the data,the capability of flake-like CeO2crystals to adsorb CO2was gradually weakening with the increase of adsorption temperature.This might be caused by an excessively high temperature resulting in the flakelike CeO2crystals being sintered and the basic sites losing their activity.
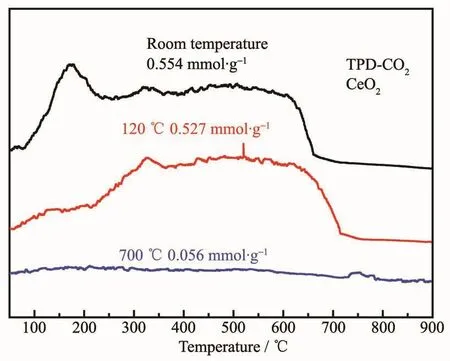
Fig.13 CO2-TPR profile of as-prepared flake-like CeO2 crystals
3 Conclusions
The flake-like CCPs were prepared by the stirring method,and the flake-like CeO2crystals were prepared by further calcining the flake-like CCPs at 500℃for 4 h.The preparation conditions were finally confirmed as follows:0.1 g CO2SM with 50 mL 0.03 mol·L-1Ce3+aqueous solution,at room temperature and 1 000 r·min-1for 0.5 h.The CO2SM could not only provide CO32-but also as a dispersant and structure directing agent.A large amount of the flakelike CCPs could be quickly prepared at room temperature.The flake-like CeO2crystals had good reduction performance and 0.554 mmol·g-1,CO2adsorption at room temperature.This study not only proposed a new method for the synthesis of metal oxide,but also contributed to investigate the ability to adsorb CO2.
Acknowledgments:This work was supported by Key Laboratory of Coal-based CO2Capture and Geological Storage(Jiangsu Province,China University of Mining and Technology,Grant No.2016A06),the National Natural Science Foundation of China (Grant No.21666027),the Program for Grassland Excellent Talents of Inner Mongolia Autonomous Region,the Natural Science Foundation of Inner Mongolia Autonomous Region (Grant No.2016JQ02)the Inner Mongolia Science and Technology Key Projects,and training plan of academic backbone in youth of Inner Mongolia University of Technology.
猜你喜欢
儿童故事画报(2021年7期)2021-10-04
内蒙古统战理论研究(2021年1期)2021-06-09
火炸药学报(2021年2期)2021-05-06
电子产品世界(2021年4期)2021-02-09
中国对外贸易(2019年4期)2019-06-26
山东工业技术(2017年17期)2017-09-13
草原(2017年8期)2017-09-13
旅游(2015年9期)2015-01-07
中国火炬(2012年1期)2012-07-24
