
甘蓝型油菜ALS基因启动子克隆及其瞬时表达分析
2019-01-09 06:51熊冬琴黄子栋张洁夫浦惠明
江苏农业科学 2018年23期
熊冬琴, 黄子栋, 彭 琦, 张 维, 张洁夫,3, 浦惠明,3, 陈 松,3
(1.江苏省农业科学院经济作物研究所,江苏南京 210014; 2.农业部长江下游棉花与油菜重点实验室,江苏南京 210014;3.江苏省现代作物生产协同创新中心,江苏南京 210014;4.江苏农林职业技术学院,江苏句容 212400)
油菜是世界上最主要的油料作物之一,也是我国重要的油料作物。我国油菜面积、总产量约占世界的1/3,是最大的油菜生产国。油菜油约占我国食用油市场的50%,是我国最主要的食用植物油。鉴于油菜在油料作物中的重要地位,油菜科学的研究一直受到学者们的高度重视。过去几十年来,油菜遗传改良已经取得显著成果,高产、优质双低油菜品种在生产上已经占据主导地位。近几年来,随着现代生物技术与分子生物学研究的飞速发展,一些生物技术在油菜基础与应用研究中也得到了广泛应用,如在油菜的分子标记辅助育种、高密度遗传图谱构建、重要性状基因的遗传定位与克隆,以及基因工程改良等研究领域都取得了显著进展。
有关油菜启动子的研究近几年来不断有报道。景寅利用反向PCR方法扩增获得肌醇半乳糖苷合成酶基因(BnGOLS1)的启动子,能驱动β-葡萄糖苷酸酶(GUS)基因在油菜组织中表达[1];朱斌等用PCR扩增方法获得甘蓝型油菜MAPK7基因家族的启动子[2];王轩鹏等根据基因组序列获得甘蓝型油菜RabGDI3基因的启动子,该启动子能驱动报告基因gus仅在花药中特异性表达,是组织特异性启动子[3];阳永学等通过PCR克隆获得油菜半胱氨酸蛋白酶家族基因BnCP51的启动子序列,通过转基因功能验证该启动子仅在花蕾、花药器官中表达,也是一种器官特异性表达启动子[4];邵铁梅等基于油菜基因组信息,利用PCR方法克隆到油菜油体蛋白基因的启动子,该启动子具有种子特异性表达特性,这也是继油菜Napin启动子后的又一个油菜种子特异性表达的启动子[5];肖旦望等用PCR方法扩增获得油菜溶血磷脂酰转移酶基因(LPAT)启动子[6]。随着相关研究的进一步开展,会有越来越多的油菜启动子被发现与应用。
植物中乙酰乳酸合成酶(ALS)是催化分支氨基酸如缬氨酸、亮氨酸和异亮氨酸生物合成第一步的酶。研究发现,抑制植物细胞内的ALS活性,会阻碍支链氨基酸(缬氨酸、亮氨酸和异亮氨酸)的生物合成,从而抑制植物细胞的分裂和生长,严重的会导致植物死亡。基于这个原理,一些化学公司陆续开发出了一系列ALS抑制剂类的除草剂并在生产上广泛应用。此外,ALS基因也受到生物学家的普遍关注,一些植物或作物的ALS基因被克隆[7-8]。研究发现,ALS基因编码序列发生碱基突变,则产生对ALS抑制类除草剂的抗性[9-10]。目前在油菜基因组中已经发现的ALS基因有3个,即BnALS1、BnALS2、BnALS3[11]。有关油菜ALS基因的研究多集中在其抗除草剂的机制上,而关于ALS基因启动子的研究及应用则鲜有报道。
本研究旨在克隆油菜ALS基因的启动子区域序列,并通过转基因瞬时表达验证其启动子活性。
1 材料与方法
1.1 材料
1.1.1 生物试剂 引物由南京擎科生物科技有限公司合成;限制性内切酶系NEB公司产品(NEB Ipswich,UK);高保真DNA聚合酶5×TransStart FastPfu PCR 试剂、凝胶回收试剂盒EasyPure®Quick Gel Extraction Kit、T/A克隆载体pEASY-T1Kit均为北京全式金生物技术有限公司产品;MS培养基系青岛海博生物有限公司产品。
1.1.2 试验材料 油菜材料为江苏省农业科学院经济作物研究所选育的抗除草剂品系M342;转基因受体材料为本氏烟(Nicotianabenthamiana),由江苏省农业科学院种质资源与生物技术研究所张保龙研究员提供。
1.2 试验方法
1.2.1 启动子序列克隆与启动子分析 根据油菜的BnALS基因序列,通过美国国立生物技术信息中心(NCBI)网站(https://www.ncbi.nlm.nih.gov/)检索其相应的油菜基因组序列,提取BnALS基因编码区5′末端上游大约1 000个碱基的序列,通过专业网站(http://molbiol-tools.ca/Promoters.htm)进行启动子预测分析。然后选取上游富含启动子元件的1 000 bp左右的序列,设计特异性扩增引物,并添加限制性内切酶位点,在上游引物的5′末端添加限制性内切酶HindⅢ的酶切位点,在下游引物的5′末端添加NcoⅠ的酶切位点,引物为PF(5′-AAGCTT GTGGAGCTGATCTTACCGACCGAAC-3′)/PR(5′-CCATGGGGTTAGAGGAGAGAGAGATGATGAA-3′)。
1.2.2 启动子序列扩增、克隆、测序及分析 采用十六烷基三甲基溴化铵(CTAB)法提取油菜M342叶片的基因组DNA。以此为模板,用高保真DNA聚合酶扩增油菜BnALS基因启动子区域的碱基序列。扩增体系:10 μL 5×Trans Start FastPfu Buffer,1 μL基因组DNA,1 μL 10 μmol/L PF,1 μL 10 μmol/L PR,5 μL 2.5 mmol/L dNTPs,1 μL Trans Start FastPfu DNA聚合酶,用ddH2O补足总体积到50μL。PCR程序:95 ℃ 3 min,95 ℃ 30 s,55 ℃ 30 s,72 ℃ 1 min,35个循环;72 ℃,5 min。用琼脂糖凝胶电泳检查扩增产物,胶回收纯化1 kb左右的PCR产物并克隆到T/A载体pEASY-T1上,转化大肠杆菌DH5α,获得重组质粒pEASY-P,挑取阳性克隆送南京擎科生物科技有限公司测序。
通过Plant CARE网站(http://bioinformatics.psb.ugent.be/webtools/plantcare /html/)对克隆到的油菜BnALS基因上游序列进行生物信息学分析。
1.2.3 启动子功能验证表达载体构建 将第1.2节克隆到的启动子序列重组到pCAMBIA1304载体上GUS基因的5′端,这样就构成了由BnALS基因启动子驱动的1个完整的GUS基因表达框。分别提取克隆载体pEASY-P质粒和由笔者所在实验室保存的植物表达载体pCAMBIA1304质粒,用限制性内切酶HindⅢ、NcoⅠ双酶切质粒DNA,回收从pEASY-P质粒切下的大小约为1 000 bp的片段,通过连接酶的作用,将启动子序列重组到目标载体pCAMBIA1304的相应酶切位点上。双酶切反应体系:总体积为50 μL,其中含有5 μL缓冲液、30 μL质粒DNA溶液、2 μLHindⅢ、2 μLNcoⅠ、16 μL H2O,于37 ℃放置6 h或过夜。酶切后,通过琼脂糖凝胶电泳,回收相应的片段,进行连接反应,连接酶反应体系:总体积为20 μL,含有5 μL线性化载体DNA、5 μL启动子DNA溶液、2 μL T4连接酶缓冲液、2 μL T4连接酶、6 μL H2O,于 16 ℃ 过夜。将连接反应产物用热激法转化大肠杆菌感受态细胞DH5α,在含有卡那霉素(Kan)的LB培养基上于37 ℃培养过夜。
1.2.4 烟草转基因试验 为了进一步验证所克隆启动子的生物学功能,将构建的表达载体pCAMBIA1304-P转入农杆菌EHA105,利用叶盘转化法转染烟草。具体试验过程如下:
1.2.5 农杆菌的准备 挑取携带载体pCAMBIA1304-P的农杆菌EHA105的单菌落,接种于5 mL含有20 mg/L利福平(Rif)和50 mg/L Kan的LB液体培养基中,28 ℃振荡培养过夜。取活化过夜的农杆菌,按1 ∶50的比例稀释到含有 20 mg/L Rif和50 mg/L Kan的YEB液体培养基中,继续培养至D600 nm约为0.6~0.8。于6 000 r/min离心5 min,收集菌体,用1/2 MS液体培养基洗涤1次菌体,并将其稀释至D600 nm为0.30~0.35。
1.2.6 烟草叶盘的遗传转化 选取苗龄约为30 d的烟草无菌苗,在无菌条件下用手术刀切取大小约为0.8 cm2的烟草叶片为外植体,投入已经准备好的农杆菌菌液中,振荡侵染 5 min 后,取出并用滤纸吸干附着于叶片表面的残液,然后放在共培养基(1×MS,3%蔗糖,1%琼脂粉,2.0 mg/L BA,0.5 mg/L IAA,pH值为5.8)上的暗处,温度设为25 ℃,共培养48 h。
1.2.7 转基因烟草苗的再生 将共培养后的烟草外植体转到芽诱导培养基(配方为1×MS+3%蔗糖+2.0 mg/L BA+0.5 mg/L IAA+500 mg/L羧苄青霉素+20 mg/L潮霉素+1%琼脂粉,pH值为5.8)上进行芽诱导培养,每隔2~3周继代1次。继代用的培养基同芽诱导培养基。待不定芽长出后,且芽长为1.0~1.5 cm时,将芽切下换到生根培养基(配方为1×MS+3%蔗糖+0.5 mg/L IAA+500 mg/L羧苄青霉素+20 mg/L潮霉素+1%琼脂粉,pH值为5.8)上诱导生根。
1.2.8 转基因烟草植株的PCR鉴定 取转基因处理的再生烟草苗叶片,用CTAB法提取基因组DNA,另取非转基因烟草苗叶片DNA作为阴性对照。用特异性引物进行PCR鉴定。引物序列:PGF,5′-CGTTCACAAACTCATTCATCATCTC-3′;PGR,5′-TTTGATGCCGTTCTTTTGCTTGTCG-3′。20 μL 扩增反应体系:10 μL 2×Tag master mix,1 μL DNA模板,1 μL 引物PGF,1 μL引物PGR,7 μL H2O。反应程序:94 ℃ 4 min,94 ℃ 40 s,55 ℃ 40 s,72 ℃ 40 min,35个循环;72 ℃ 5 min,用琼脂糖凝胶电泳检测扩增产物。
1.2.9 GUS活性检测 用北京Solarbio生物科技有限公司生产的GUS染色液试剂盒定性检测GUS基因的表达活性。将转基因烟草PCR阳性单株幼苗完整取出,去除根部的琼脂,用无菌水清洗后,将植株浸泡在GUS显色液中,于室温下放置3~6 h后,用无水乙醇进行脱色处理,镜检拍照。以非转基因幼苗作为对照。
2 结果与分析
2.1 油菜ALS基因启动子序列的克隆与分析
本研究根据BnALS基因上游序列设计特异性引物,通过PCR扩增油菜基因组DNA的方法获得大小约为1 000 bp的片段,详见图1。对该片段进一步克隆、测序,实际获得长度为1 048 bp的BnALS基因上游序列(序列1),详见图2。
通过Plant CARE网站(http://bioinformatics.psb. ugent.be/webtools/plantcare/html/)对克隆到的BnALS基因上游序列进行生物信息学分析,结果显示,该序列含有与启动子相关的多种顺式作用元件,如CAAT-box、TATA-box等,表明该序列具有真核细胞启动子的结构特征。部分顺式作用元件的情况见表1。

2.2 启动子功能验证表达载体的构建
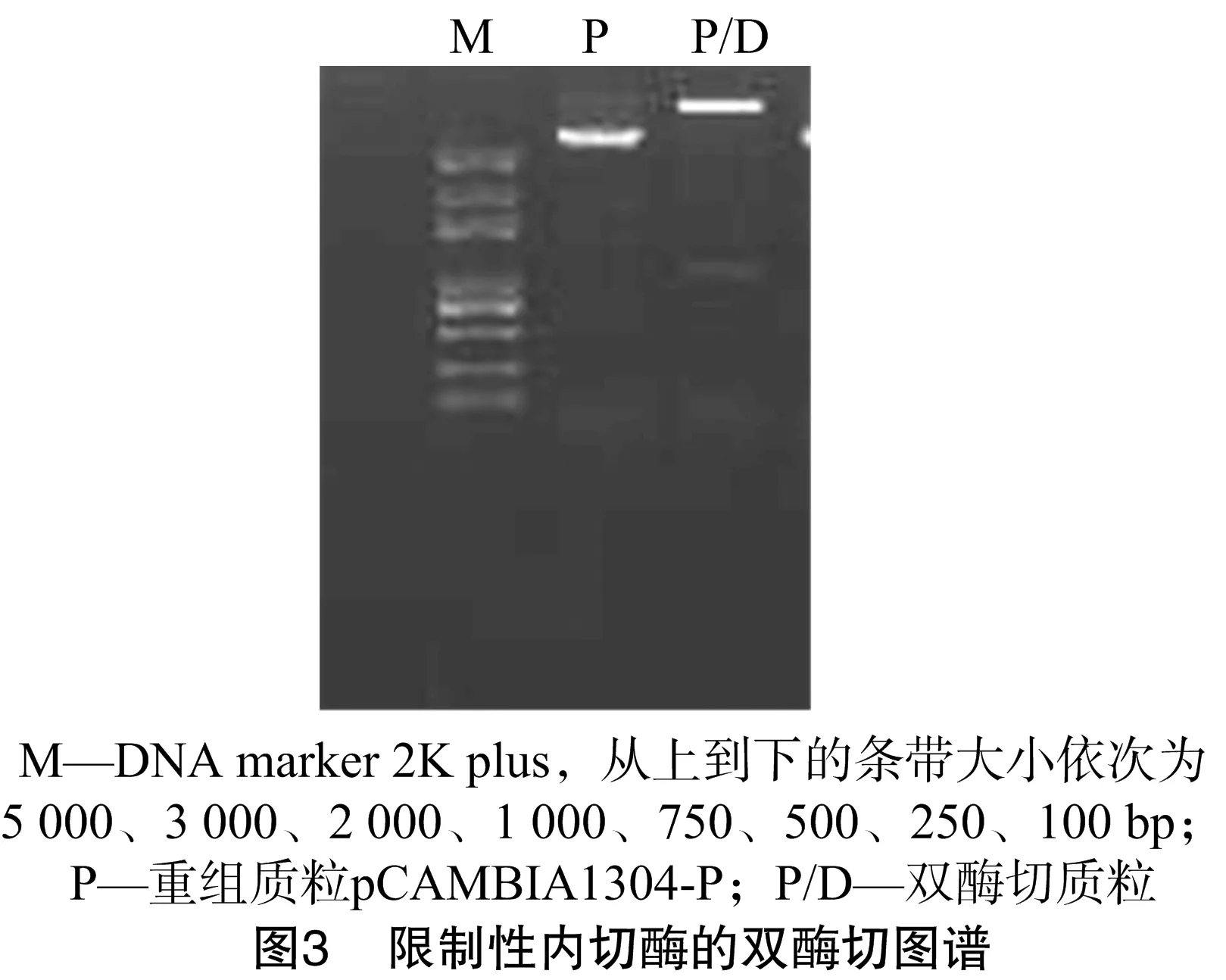
为了进一步验证所克隆的BnASL基因上游序列是否具有启动子功能,将该序列重组到pCAMBIA1304载体上GUS基因的5′-端,这样就构成了由BnALS基因的上游序列驱动的1个完整的GUS基因表达框。分别提取获得的克隆载体pEASY-P质粒和笔者所在实验室保存的植物表达载体pCAMBIA1304质粒,并用限制性内切酶HindⅢ、NcoⅠ双酶切质粒DNA,回收从pEASY-P质粒上切下的大小约为 1 000 bp 的片段,重组到双酶切线性化的pCAMBIA1304载体上,获得重组载体pCAMBIA1304-P。重组载体经限制性内切酶酶切验证,可以得到1个大小约为1kb的片段,证明启动子序列已经重组到目标载体上(图3)。经测序分析可知,所克隆的启动子序列准确插入相应的酶切位点,无缺失、错配现象,表明该重组载体可以用于转基因功能的验证工作。


表1 ALS启动子序列含有的主要相关顺式作用元件
2.3 烟草转基因试验
为了进一步验证所克隆的油菜BnALS基因上游序列的启动子功能,将构建的表达载体pCAMBIA1304-P转入农杆菌EHA105,利用叶盘转化法转染烟草。烟草叶片经过农杆菌侵染、抗性芽诱导、抗性芽生根等阶段的再生培养,最终获得转基因烟草幼苗,其过程详见图4。
2.4 转基因烟草植株的PCR鉴定
利用设计的特异性PCR引物扩增再生幼苗的基因组DNA,鉴定转基因阳性单株。由图5可以看出,经过PCR检测,有5株幼苗扩增到与质粒对照相同的阳性条带,2株幼苗未扩增到相应的条带。


2.5 GUS基因表达活性检测
以PCR结果为阳性的转基因再生烟草幼苗及非转基因处理的烟草幼苗对照,用GUS显色试剂盒进行GUS基因表达活性的定性检测。将烟草幼苗完整取出,浸泡在GUS基因显色液中,于室温下放置3~6 h后,用无水乙醇脱除背景色,如果有GUS基因表达活性,组织就会出现蓝色反应。结果显示,转基因烟草全株均出现明显的蓝色反应,而非转基因幼苗则呈淡淡的黄色,转基因烟草GUS活性定性检测结果见图6。以上结果表明,本研究克隆的BnALS基因上游序列具有较强的组成型表达活性, 能驱动报告基因在烟草幼苗各个器官中的表达。

3 讨论
与其他真核生物一样,植物启动子区域最具特征的就是TATA box序列,这是RNA聚合酶Ⅱ识别的位点,也是一些反式作用因子与DNA相互作用的位点之一。TATA box与转录起始点之间的碱基长度是转录精确起始的必需因素,植物启动子的TATA box多在转录起始点上游(32±7) bp处[12]。TATA box通过与转录因子TFⅡD的识别结合而发挥功能,决定RNA聚合酶起始转录的位点,以及介导前转录复合物的形成并起始转录;起始因子(initiator,简称Inr)是基因启动子核心结构的第2种类型,与转录起始位点重叠[13]。Inr元件并无十分严格的同源顺序,其中最关键的核苷酸是处于+1位置的A和+3位置的T。Inr在功能上与TATA框类似,常通过与TFIID的结合决定转录起始点的位置起始转录,并能介导上游至少一部分激活因子的调控作用;CAAT box是Shirsat等于1989年在豌豆legA基因启动子中发现的1个增强转录的元件,一般位于-75 bp附近,其保守序列为GGC/TCAATCT[14]。CAAT box控制着转录起始的频率,其对基因转录的激活作用存在双向性,且作用距离不固定;GC box通常位于-90 bp附近,保守序列为GGGCGG,可有多个拷贝,并能以任何方向存在而不影响其功能。Kuwahara等研究发现,GC box需要与另一特异的转录因子(SP1)结合才能促进基因转录[15]。植物启动子的顺式元件除了CAAT box和GC box以外,不同来源的启动子通常还具有种属特异的或者仅局限于某种基因家族特有的调节序列。
本试验根据油菜BnALS基因上游序列设计特异性引物,通过PCR扩增获得的ALS基因上游的1 048个碱基序列中包含多个TATA box和CAAT-box启动子核心序列,仅在正义链上就检测到11处TATA box序列和3个CAAT-box,这些启动子的核心序列可能决定了转录的起始、方向和转录效率。根据植物基因转录起始位点通常与上游TATA-box 的距离为(32±7) bp,以及转录起始碱基多为A碱基的规律,推测本试验克隆的启动子起始位点可能是位于1 026 bp或 1 028 bp 处的A碱基。此外,BnALS基因上游区域还含有其他多种转录调控元件,主要有激素响应元件、脱落酸响应元件ABRE-motif、甲基茉莉酸响应元件CGTCA-motif和生长素等激素响应元件TGA-element,说明ALS基因在激素响应过程中可能起作用。此外,还有非生物胁迫响应元件HSE和MBS,因此可见,ALS基因也可能参与干旱或热激反应的应答。另外,还发现有多个光响应元件,暗示ALS基因的表达可能还受到光的调控。
人们研究启动子的目的主要是为了弄清一些结构基因的表达与调控模式,除此以外,启动子在植物基因工程研究领域也有重要的应用价值。启动子对外源基因的表达水平影响很大,是基因工程表达载体的重要元件。目前在植物基因工程中应用最多的启动子是来自花椰菜花叶病毒(CaMV)的35S启动子和农杆菌胭脂碱合酶基因的nos启动子[16]。这2个组成型表达的启动子常用于双子叶植物基因工程,而在单子叶植物中应用较多的是来自玉米的Ubi1启动子[17]和水稻的Act1启动子[18]。人们不断开发研究新启动子应用于植物的基因工程,目的是为了在植物中更高效地表达目的基因。重复使用同一种组成型启动子驱动2个或2个以上的外源基因表达可能会引起基因沉默或者发生共抑制[19]。另外,将从病毒基因组中克隆的启动子序列应用到植物基因工程中,可能会引起人们对转基因植物生物安全性的担忧,因此,从植物本身克隆活性强的组成型启动子也就成了植物基因工程研究的一个重要方面。Shirasawa-Seo等从拟南芥中克隆了色氨酸合成酶亚基(PTSB1)和植物光敏色素B基因的启动子(PPHYB),替代CaMV35S的组成型启动子,二者分别与GUS基因融合表达,活性甚至高于35S启动子,驱动NPTⅡ基因表达,具有相同的效果,可以认为拟南芥的PTSB1和PPHYB启动子是植物来源的可替代35S启动子的强组成型启动子[20]。本试验克隆到了BnALS基因启动子序列,通过生物信息学软件预测了启动子元件组成,并构建了与GUS基因融合的植物表达载体,通过转基因烟草的瞬间表达分析,初步结果显示,所克隆的启动子序列能驱动GUS基因在烟草幼苗多个器官中表达,表明该序列具有较强的组成型启动子的表达活性。
猜你喜欢
学与玩(2022年10期)2022-11-23
今日农业(2022年3期)2022-06-05
今日农业(2021年21期)2021-11-26
今日农业(2021年14期)2021-10-14
娃娃乐园·综合智能(2018年3期)2018-03-22
数学小灵通(1-2年级)(2017年10期)2017-11-08
现代工业经济和信息化(2016年2期)2016-05-17
创新科技(2015年1期)2015-12-24
电子工业专用设备(2015年4期)2015-05-26
无机化学学报(2014年3期)2014-02-28
