
民猪全基因组序列测定与分析
2018-12-21 03:48张冬杰何鑫淼王文涛
东北农业大学学报 2018年11期
张冬杰,何鑫淼,王文涛,汪 亮,刘 娣*
(1.黑龙江省农业科学院畜牧研究所,哈尔滨 150086;2.农业农村部种养结合重点实验室,哈尔滨 150086)
民猪是我国优良地方猪种,具有高繁殖、耐粗饲、抗逆、肉质优良等特点,但生长速度慢、瘦肉率低、料肉比高。民猪是约300年前由河北和山东的大量移民经陆路迁至辽宁西部的小型华北黑猪及海路迁至辽宁南部和中部的山东中型华北黑猪与当地猪种杂交后,经多年选育而成[1]。文献记载,旅顺郭家村发掘的晚期龙山文化遗址(距今约五千年)及黑龙江省宁安县镜泊湖南端的莺歌岭遗址(距今约三千年)[2]中均发现原始社会陶猪,体态与野猪区别明显,说明东北地区在新石器时代晚期已开始驯养野猪。因此,现存民猪具有华北黑猪和黑龙江省本地猪血统。但受技术条件所限,多年来仅通过表型鉴定区分猪种,无法从遗传学角度深入分析。
随着二代测序技术发展,可从全基因组水平分析和探讨物种分子进化、基因组成和基因调控规律[3]。Li等测定藏猪群体全基因组序列发现,藏猪与家猪祖先曾向不同方向进化[4]。Ma等通过荣昌猪、松辽黑猪、大白猪和长白猪全基因组扫描发现,受选择区域占常染色体3.74%~5.33%,与繁殖、毛色和耳型性状相关的多个基因受选择[5]。Wang等通过600头大白猪和600头长白猪全基因组扫描发现,与脂肪酸合成、生长发育、免疫应答等相关基因受选择[6]。
为深入分析民猪遗传背景,探讨民猪优良种质特性遗传基础,有效保护和利用民猪资源,本研究以小兴安岭东北野猪为参照,利用Illumina Hiseq2000测序平台,采用双末端测序方法,测定10头民猪和4头东北野猪全基因组序列,从分子水平探讨民猪遗传背景及基因组选择情况。
1 材料与方法
1.1 试验材料
选择10头无血缘民猪(二民猪),其中公猪4头,母猪6头,均来自黑龙江省农业科学院畜牧研究所民猪养殖基地。东北野猪4头,均为母猪,来自黑龙江省小兴安岭林区。
1.2 方法
1.2.1 DNA提取与检测
分别采集14头个体耳组织样,按照常规苯酚抽提方法提取基因组DNA。采用琼脂糖凝胶电泳结合紫外分光光度计检测方法,测定DNA样品浓度,确保每个样品DNA浓度不低于200 ng·μL-1,DNA总量不低于500 μg,OD值介于1.8~2.0。电泳检测无明显RNA条带,基因组条带清晰、完整,主带应在100 kb以上。
1.2.2 原始数据过滤、整理及质量评估
按照Illumina公司操作手册构建14个插入片段长度在425~448 bp之间文库,利用Illumina Hiseq2000测序平台作双末端2×100 bp测序,对所获原始fastq格式文件作过滤和比对(mapping),过滤标准为平均质量分数>=Q30;总“N”碱基数< 3;5′端前50 bp无“N”碱基。
1.2.3 基因组重测序分析
从Ensembl数据库(http://www.ensembl.org/Sus_scrofa/Info/Index)中下载参考基因组序列(Sscrofa 10.2)。采用bwa(0.7.5a-r416)aln程序分别将经过滤后Clean reads数据比对至参考基因组序列中,均选择默认参数。主要步骤包括:①对参考基因组构建索引;②寻找输入reads文件的SA坐标;③生成sam格式比对文件,若一条read比对到多个位置,则随机选择一种。比对结束后,重新排序sam文件,利用Samtools(0.1.19)工具包将其转换成bam文件格式,picard1.81软件对bam文件排序。
1.2.4 SNPs分析
使用GATK基因组分析工具箱鉴定样品SNPs,具体步骤如下:①将上述经排序bam文件采用GATK(v2.2-14-g11728e9)程 序 中 Realigner TargetCreator命令重新比对所有Reads,提高预测准确率;②采用UnifiedGenotyper工具获取SNPs位点,stand_call_conf设置为30,stand_emit_conf设置为10.0;采用Variant Filtration程序过滤上述所得SNP位点,将满足以下任一条件SNPs滤掉:FS>60,HaplotypeScore>13.0,MQ<40,QD<2,MQRank Sum<-12.5,ReadPosRankSum<-8.0,dp<4或dp>50,10 bp内包含3个或以上SNP。过滤结束后统计SNPs位点,采用Ensembl提供工具包(Variant Effect Predictor)注释SNPs。
1.2.5 InDel分析
采用 UnifiedGenotyper(stand_call_conf设置为30,stand_emit_conf设置为10.0)命令获取InDel信息,过滤InDel,将满足以下任一条件InDel滤掉:QD<2.0,ReadPosRankSum<-20.0,FS>200,dp<4或dp>50。过滤结束后统计InDel位点,采用Ensembl提供工具包(Variant Effect Predictor)注释InDel。
1.2.6 群体遗传分析
从NCBI数据库中下载50个不同地区家猪和野猪全基因组序列,采用上述相同方法分析。使用EIGENSOFT5.0.2软件包中smartpca软件[7],基于常染色体上SNPs信息作主成分分析(Principle Component Analysis,PCA)。使用MEGA 5.0软件[8]构建基于NJ算法不同猪种间分子进化树。
1.2.7 选择性清除分析
应用滑动窗口方法(500 kb窗口以50 kb步滑动)计算民猪和东北野猪群体间多态性水平(θπ)和遗传分化(Fst)。受选择区域应同时满足显著低或高θπ比率(5%左右尾,θπ比率分别是0.659和1.770)以及显著高Fst值(5%右尾,Fst值是0.364)。
2 结果与分析
2.1 原始数据处理与比对
10头民猪平均获得36.91 Gb原始数据,经质量过滤后获得31.29 Gb干净数据,平均测序深度为7.76×,其中85.46%数据比对到参考基因组,1×测序覆盖率至少为97.90%,4×测序覆盖率至少为85.95%。4头东北野猪平均获得43.51 Gb原始数据,经质量过滤后获得36.28 Gb干净数据,平均测序深度为8.91×,其中86.27%数据比对到参考基因组,1×测序覆盖率至少为97.92%,4×测序覆盖率至少为87.43%。
2.2 SNP检测及注释结果
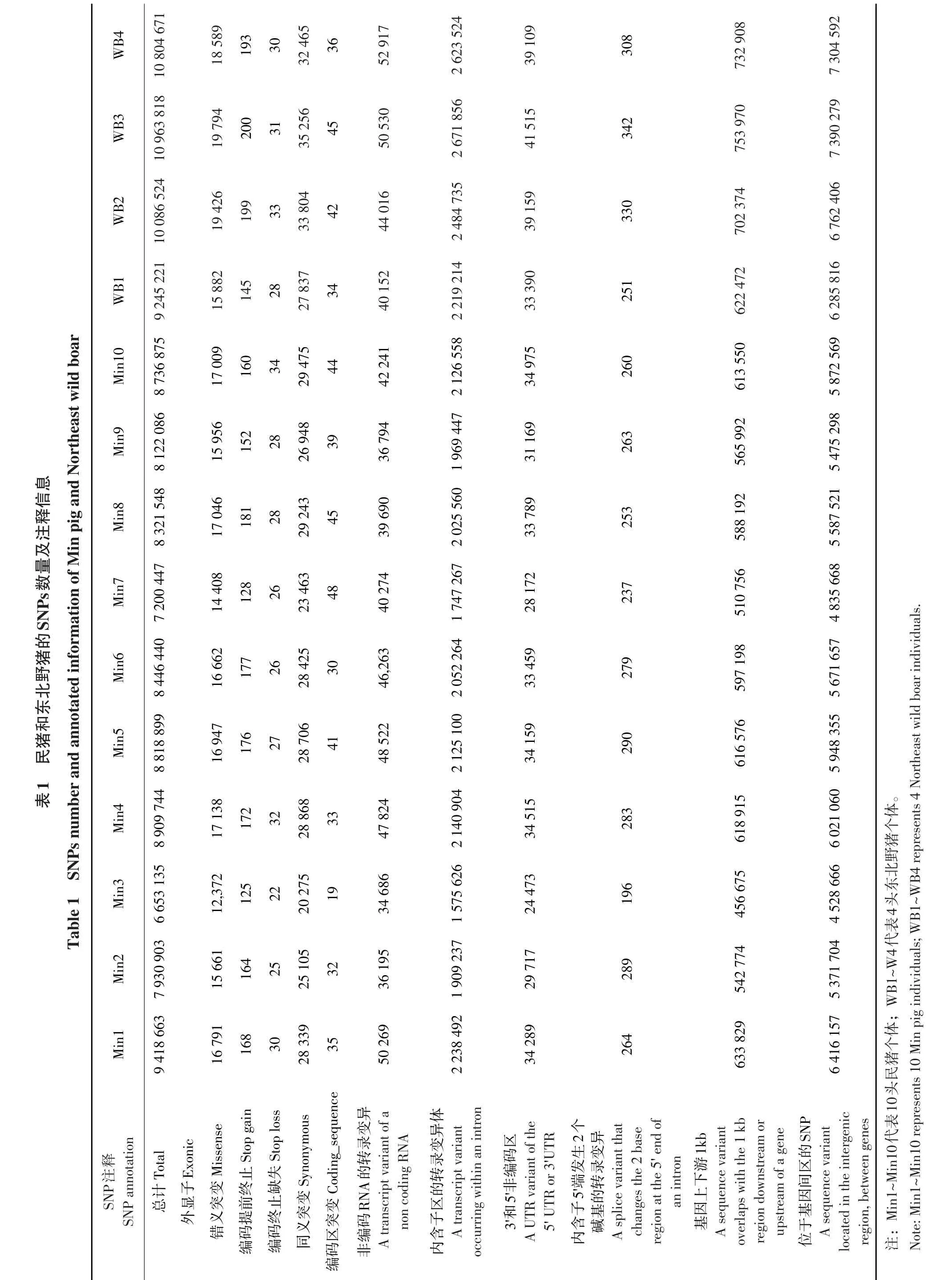
10头民猪平均检测到8 255 874个SNPs;注释后发现,发生错义突变SNPs数量平均为15 999个,占总数(8 255 874)的0.19%;发生同义突变SNPs数量平均为26 885个,占总数0.33%,可见基因组内同义突变比率明显高于错义突变。位于外显子区SNPs数量平均为43 108个,占总数0.52%。大量SNPs发生在基因间区及内含子区,分别为5 572 866个和1 991 046个,占总数67.50%和24.12%。SNP在各条染色体上分布情况见图1。1号染色体上SNP数量最多,其次是13号染色体,Y染色体最少,仅为2227个。
4头东北野猪基因组内平均检测到10 275 059个SNPs,发生错义突变SNPs数量平均为18 423个,占总数(10 275 059)0.18%;发生同义突变SNPs数量平均为32 341个,占总数0.31%。位于外显子区SNPs数量平均为51 017,占总数0.50%。大量SNPs同样发生在基因间区及内含子区,分别为6 935 773个和2 499 832个,占总数67.50%和24.33%,各类SNPs所占比率与民猪基本相同。民猪和东北野猪SNPs数量及注释信息见表1。
2.3 民猪和东北野猪与已知猪种SNPs信息比对
将本研究中获得10头民猪和4头东北野猪SNPs信息与包含138头猪的dbSNP数据库比对发现,有19 469 459个SNPs与dbSNP数据库中信息一致,占dbSNP数据库总数68.06%,但有7 739 173个为新发现SNPs,可补充猪SNPs数据库信息。
2.4 InDel检测及注释结果
10头民猪平均检测到739647个Indel位点,其中相对于参照染色体纯合InDel位点451099个,杂合InDel位点288548个;InDel位点注释后发现,所有染色体上纯合均多于杂合InDel位点,1号染色体上InDel位点最多,共计112 419个,占总数(739 647)15.20%,其次是13号染色体,共计81687,占总数11.04%,8号和2号染色体上InDel数量相近,分别为69 792个和69 548个。性染色体Y上InDel位点最少,仅151个。民猪InDel在各条染色体上分布情况见图2。
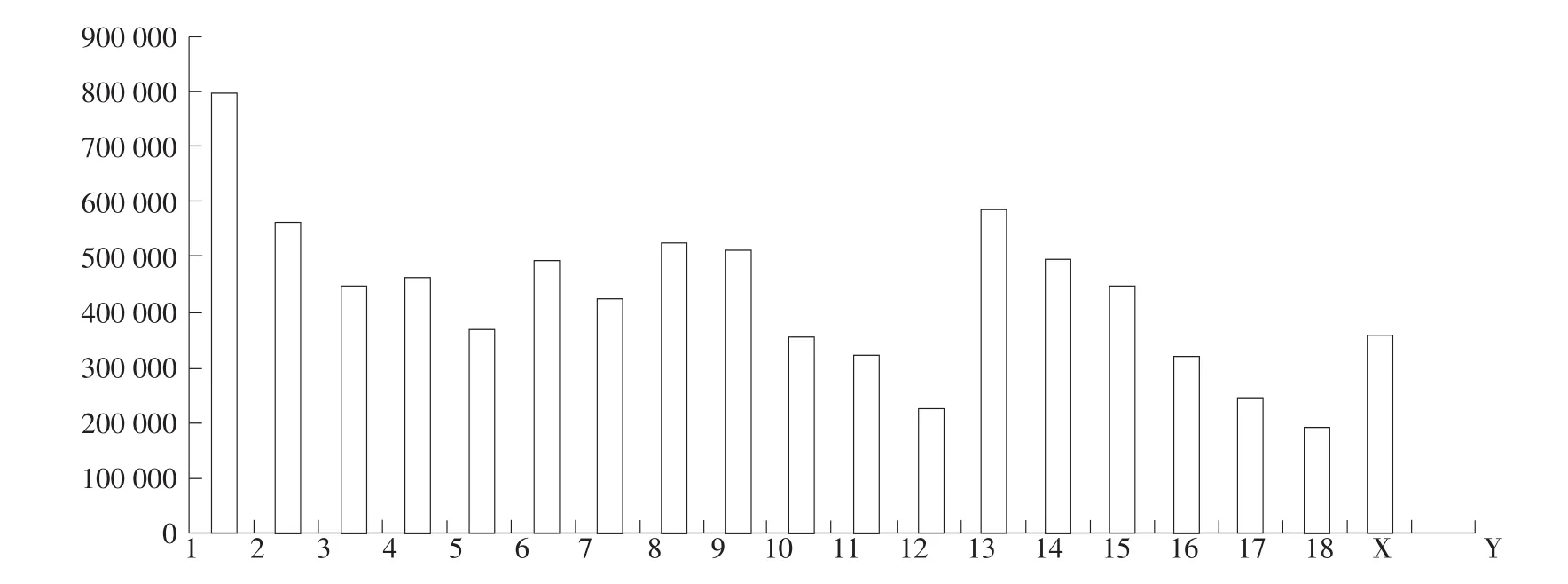
图1 民猪SNPs在染色体上分布情况Fig.1 Distribution of SNPs on chromosome in Min pig
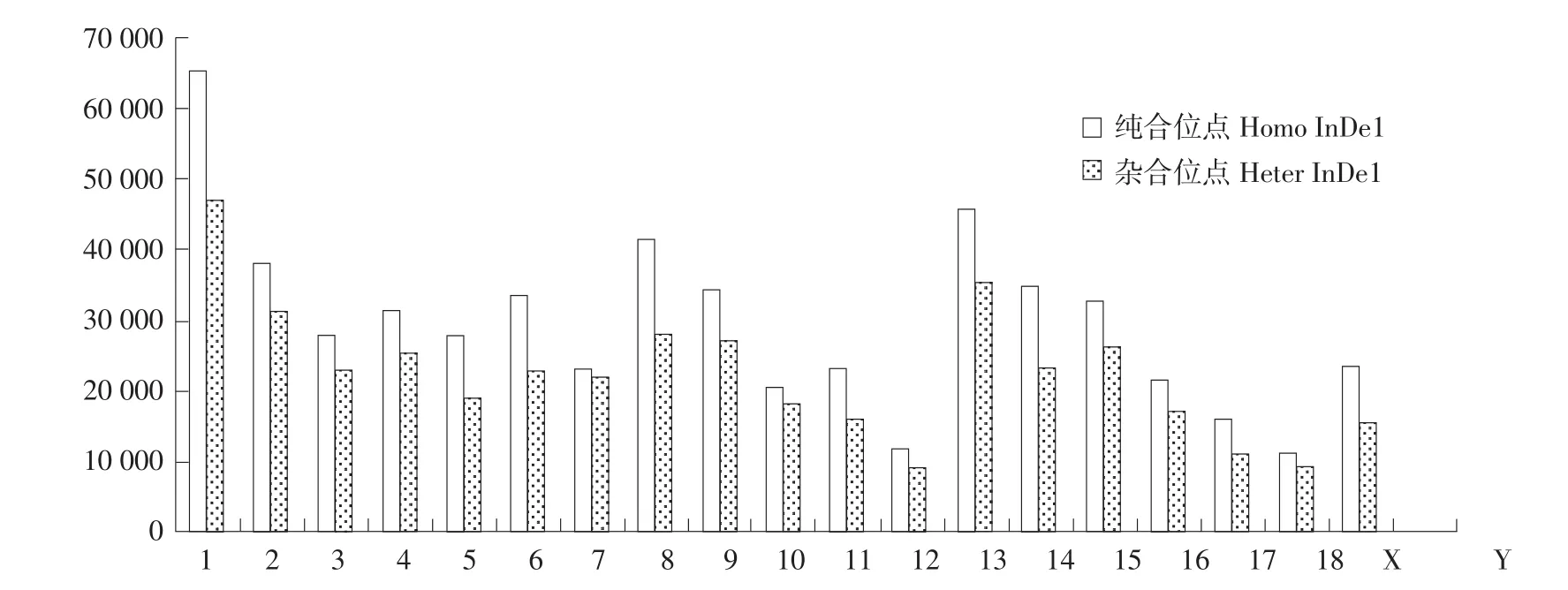
图2 民猪InDel在染色体上分布情况Fig.2 Distribution of InDel on chromosome in Min pig
4头东北野猪平均检测到1265842个Indel位点,其中相对于参照染色体纯合InDel位点781299个,杂合InDel位点484542个。所有染色体上纯合均多于杂合InDel位点,1号染色体上InDel位点最多,共计133841个,占总数10.57%,其次是13号染色体,共计112027,占总数8.85%,8号和2号染色体上InDel数量相近,分别为84799和84000个。性染色体Y上InDel位点最少,仅181个。东北野猪InDel在染色体上整体分布趋势与民猪基本一致,但数量高于民猪。
2.5 主成分分析结果
利用NCBI数据库中下载的包括欧洲家猪、中国家猪、亚洲野猪、欧洲野猪和疣猪5个类群共计50个SNPs数据(见表2),结合本研究中测定10头民猪和4头东北野猪SNPs数据,作主成分分析。通过主成分分析发现(见图3),猪种聚类情况与其地理分布基本一致,疣猪属于野猪一种,主要分布在非洲,与欧洲和亚洲猪种亲缘关系较远,在PCA图中与其他猪种距离最远。欧洲家猪和欧洲野猪距离最近,亚洲家猪和亚洲野猪距离最近,东北野猪与亚洲家猪、野猪聚为一类,但民猪处在欧洲和亚洲猪种之间,说明民猪在品种形成过程中曾引入欧洲猪血统。
?
表2 不同猪种NCBI登录信息Table 2 NCBI login information of different pig breeds
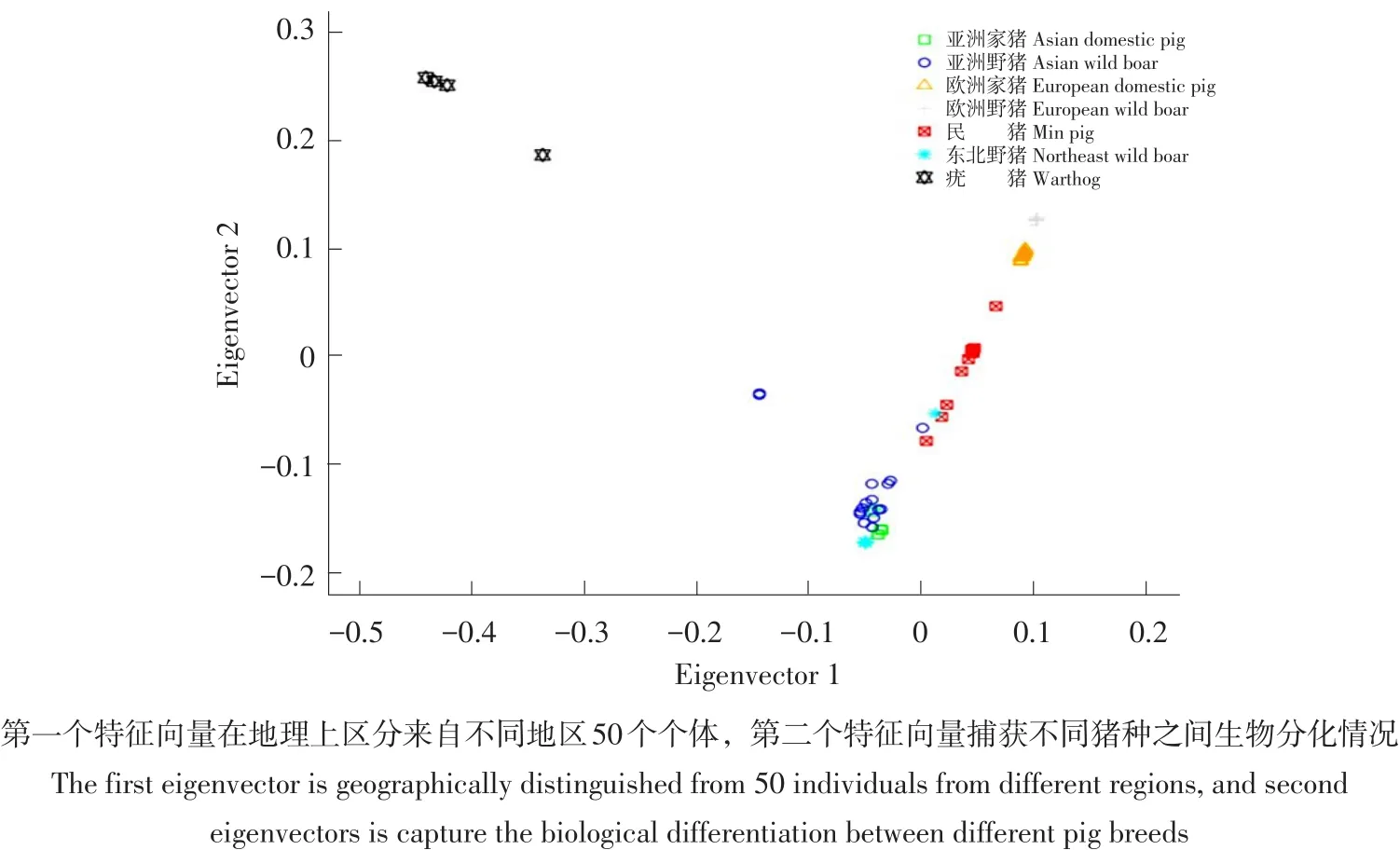
图3 主成分分析结果Fig.3 Principal component analysis results
2.6 分子进化树
基于NJ算法构建分子进化树与主成分分析结果基本一致,在亲缘关系上,民猪居于亚洲和欧洲猪种之间,但群体内存在遗传分化;东北野猪与中国北部野猪和日本野猪遗传距离较近。具体聚类结果见图4。
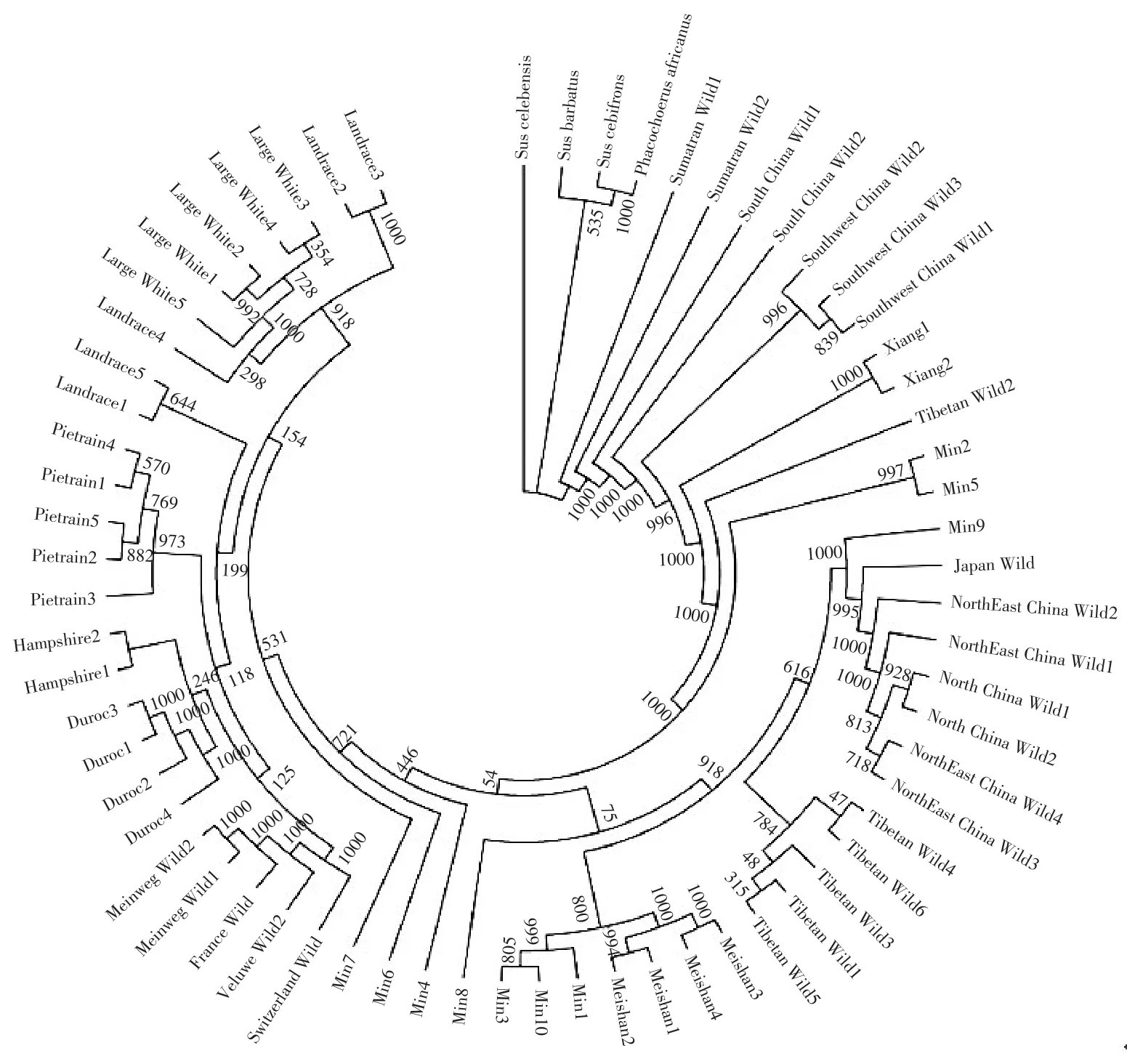
图4 分子进化树Fig.4 Molecular phylogenetic tree
2.7 选择性清除分析
同时使用θπ和Fst两个参数筛选民猪和东北野猪基因组内受选择基因(见图5)。民猪基因组内有15.71 Mb区间受选择,占总基因组0.559%,包含181个基因,其中有功能注释基因118个。东北野猪基因组内有29.81 Mb区间受选择,占总基因组1.061%,包含411个基因,其中有功能注释基因279个。

图5 Fst和θπ选择性清除分析Fig.5 Selective clearance analysis of Fst and θπ
民猪基因组中受选择基因包括与肌节中能量转移相关的肌酸激酶线粒体2基因(CKMT2);作为动力蛋白与细胞骨架相关肌球蛋白基因(MYO1C);与脂质转运相关脂质运载蛋白9和15(LCN9和CN15),载脂蛋白A-V(APOA5);与过氧化物酶合成有关过氧化物酶体的生物合成因子1(PEX1);与脊椎发育相关VRTN基因(VRTN);与视神经发育相关视神经萎缩基因(OPA1);与脂类代谢相关促肾上腺激素释放激素2(CRHR2)等。野猪基因组中受选择基因包括与神经系统有关嗅觉受体基因(OR2B6,OR8b8和OR8b4)、光传感因子3(PDCL3)和激素肽3(TAC3);与免疫相关NLR基因家族(NLRP4和NLRP11)、干扰素-欧米伽2(IFNOMEGA-2)和叉头框蛋白1(Foxn1);与免疫球蛋白相关破骨细胞受体基因(OSCAR);与雄性生殖相关羟甾类脱氢酶6(HSD17B6)、精子酵素结合蛋白(ACRBP)、附睾精子结合蛋白1(ELSPBP1)和鱼精蛋白(PRM1)等;与线粒体能量代谢相关细胞色素C氧化酶(COX)和NADH脱氢酶(NDUFA9)等。
3 讨论
猪是最早被驯化的家养动物之一,分别在亚洲和欧洲独立驯化。中国位于亚洲驯化中心,拥有黄河中部和东北部两个不同起源地[9]。从17世纪开始,中国部分地方猪种和欧洲猪种发生杂交[10]。为提高生产性能,中国本地猪种之间开展杂交育种,一些有利等位基因在不同猪种间交流并固定[11]。因此,中国地方猪遗传背景比其表型更为复杂。
地方猪亲缘关系和遗传距离早期分析主要依靠线粒体基因组序列或微卫星[12]。随着二代测序技术成熟,从全基因组水平筛选SNP标记并作群体遗传分析,准确度和可信度提高。本研究检测到SNPs有68.06%与dbSNP数据库中一致,但仍有7 739 173个新发现SNPs,既可补充现有SNP数据库,同时也是分析民猪和东北野猪种质特性遗传基础。统计筛选到SNPs在染色体上分布情况发现,SNPs数量与染色体长度呈正相关,与前人研究结果相近[13]。进一步分析发现,无论是SNPs还是InDel数量,东北野猪均高于民猪,表明野猪群体与家养民猪相比,具有丰富多样性。可能因东北野猪为野生群体,未受高强度人工选择影响。
基于全基因组SNPs信息构建的分子进化树,可解决前期每个基因代表不同进化历程、基因横向转移等造成的不同基因构建进化树不同的问题[14]。本研究构建分子进化树及主成分分析结果均清晰显示,民猪在进化关系上介于亚洲猪种和欧洲猪种之间,东北野猪与我国北方野猪及日本野猪聚于一个较大分支,与其地理分布位置相符。民猪含有部分欧洲猪血统,推测与黑龙江省地理位置相关,与俄罗斯存在悠久商贸历史。据《黑龙江省志》介绍,20世纪20年代(1914~1931年),黑龙江省内养猪已发展到126.2万头,包括民猪及中东铁路沿线苏白猪,苏白猪含部分英国大白猪血统。Ai等探讨不同猪种间杂交现象时发现,猪X号染色体上存在长达14 Mb低重组区,南北地方猪在该区域存在两种不同单倍型,北方猪单倍型可能来自另一个已经灭绝猪属,该属间杂交事件据推算发生于数十万年前[15],说明民猪除含有山东中型华北黑猪及欧洲猪血统外,还包含黑龙江省本地古老猪种血统。由此可见,民猪遗传背景相对复杂,遗传资源宝贵。
选择性清除分析发现,民猪与东北野猪相比多个与脂类代谢相关基因受到选择,主要由于早期“肥猪”选育目标,造成与大白猪基因组中肌肉生长发育相关基因受到强烈选择[16]。此外,育种者通过增加体长获得更高产肉量,通过表型长期选择,与脊椎发育相关VRTN基因受到选择[17]。由于家猪祖先常夜间活动,视力较差,听觉与嗅觉灵敏。在自然选择作用下,决定视神经发育OPA1基因[18]受到选择。东北野猪作为野生动物比家养动物更适应复杂自然环境,躲避天敌、抵抗疾病等,导致其基因组内受选择基因数量多于民猪,与神经、免疫、生殖等性状相关多个基因均受到选择。Groenen等研究表明,家猪与野猪相比,参与免疫反应和嗅觉感应的基因快速进化[9]。利用基因组重测序策略鉴定遗传变异往往因参考基因组序列不完整而存在一定局限性,本研究使用猪10.2版本参考基因组序列来源于一只杜洛克家猪,一些民猪特有遗传信息因无法比对而缺失。今后应采用de novo组装策略,尽快建立并完善我国地方猪基因组信息,发现更多新变异及新序列、基因。
猜你喜欢
猪业科学(2022年10期)2022-11-03
小天使·一年级语数英综合(2022年3期)2022-04-16
汉语世界(2021年6期)2021-12-17
汉语世界(The World of Chinese)(2021年6期)2021-12-13
猪业科学(2021年6期)2021-08-12
猪业科学(2021年3期)2021-05-21
意林(2021年3期)2021-03-11
智慧少年·故事叮当(2019年2期)2019-06-11
小学阅读指南·低年级版(2017年6期)2017-06-12
读写算(下)(2015年6期)2015-08-22
