
美国食品药品管理局2017年终总结与2018展望
2018-12-19 10:21美国食品药品管理局局长ScottGottlieb王培力
中国食品药品监管 2018年2期
文 / 美国食品药品管理局局长Scott Gottlieb 译 / 王培力
本文是美国食品药品管理局局长Scott Gottlieb在2017年底发表的年终总结与新年展望,从五个方面总结性回顾了过去一年FDA在食品药品监管领域取得的显著成绩,并表示2018年FDA将以确保创新转化为惠及公众的应用成果作为重要目标。
如今,医药领域的突破革新正在以前所未有的速度深刻地改变着我们研究和治疗疾病的方式。在此背景下,食品药品管理局(Food and Drug Administration,FDA)力求评估和改善各方面政策,以确保在保护消费者的前提下,促进有益创新,更有效地治疗人畜疾病,改善公共健康。本文中FDA局长Scott Gottlieb博士回顾并总结了2017年FDA在医药和公共卫生方面的经验,为开展2018年的工作提供参考。
2017:FDA创新审批破纪录
随着人们对疾病认识的不断提高,医学实践也越来越符合患者的个性化需求。如何在发展过程中与创新者合作,使用最好的科技手段高效地为患者提供医疗产品,是我们的不懈追求。例如,FDA最近批准了一种新型基因诊断设备,该设备使得医疗服务中心的单次测试即可以检测到多个甚至数百个基因突变位点,使得早期诊断成为可能。
此外,在迅速发展的个体化医学领域,FDA草拟了个性化医学指导方案(以下简称“方案”),改善潜在变化致病的分子,特别是罕见病分子(如基因突变)的治疗方法。当科学实验有效证明某种药物可能对特定基因组的患者有效时,药物开发者便可基于对罕见病基因突变的鉴定将患者纳入临床试验中进行靶向治疗。方案还讨论了在某些疾病中存在的有待证明的各种分子亚型,以更加科学地规范和指导个体化、针对性治疗。
2017年8月,FDA批准了美国首个基因治疗产品,这标志着以一种全新的方式治疗疾病的时代已经到来。目前,已有两种基因治疗药物获得批准,这在基因治疗领域具有重要里程碑意义,也给治愈遗传病和顽疾等严重性疾病带来了希望。
其他方面的创新也取得了一些类似的历史性成就。在开发过程早期识别目标患者,更及时有效地将靶向药物作用于疾病的潜在机制,这些进步反映了科学研究的巨大进步,为我们在新的平台攻克更多的疾病创造可能。
得益于FDA已取得的这些成就, 2017年FDA共批准了56种新型药物和生物制剂,其中46个是由药物评估和研究中心批准的新分子实体(包括28个使用一个或多个FDA快速审查计划批准的药物);另外10个是由生物制剂评估和研究中心批准的生物制剂(如图1所示)。仿制药(Generic Drugs)的获批数量多次达到最高月纪录,年度总数(1027种)也创历史新高(如图2所示)。此外,孤儿药(用于治疗罕见疾病的药物)获批数量创历史新高。与此同时,我们完成了所有孤儿药认证申请积压的工作。以目前的趋势,2018年仿制药物获批数量将超过2017年。
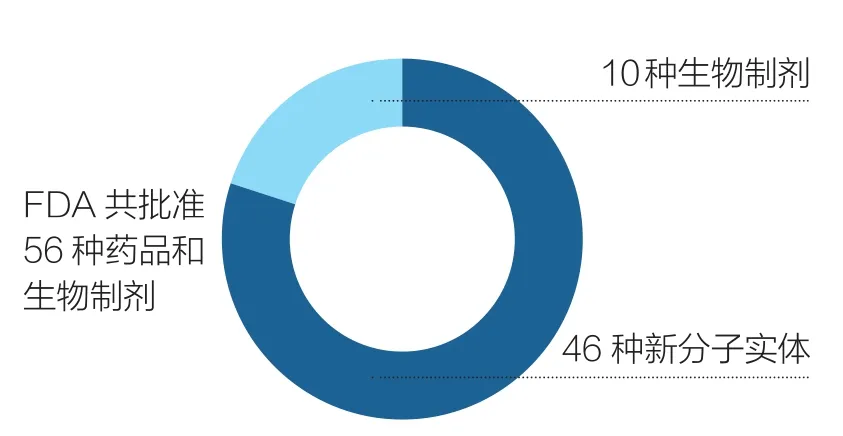
图1 2017年FDA创新药物和生物制剂审批情况
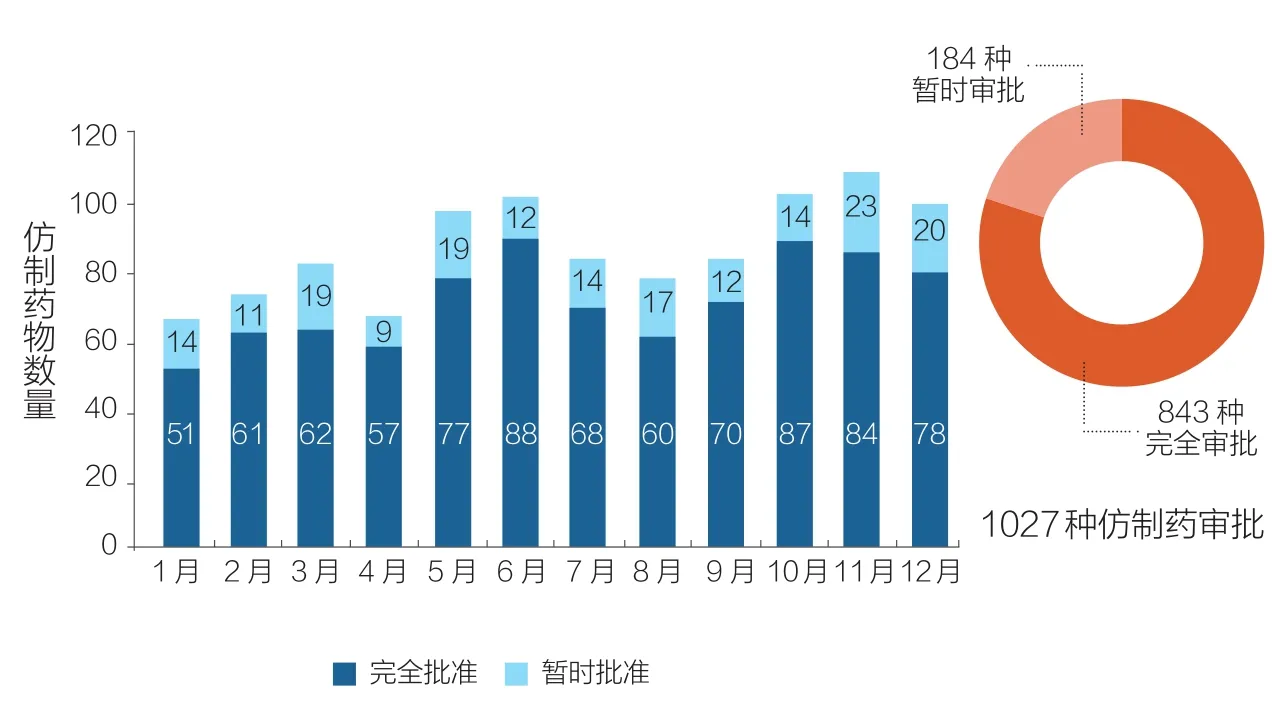
图2 2017年FDA仿制药审批情况
以科学为基础,以患者为中心的监管方式也延伸到了医疗器械领域。以产品开发的生命周期为基础,简化临床开发协议,缩短上市前的研究、审批时间,并通过上市后全面广泛地收集临床数据等举措,既减少了患者的等待时间,又依据这些证据增加了审批的科学性和权威性。这种灵活的举措使得医疗器械生产企业更加严格地执行FDA的标准,保证了产品的安全性和有效性。
运用“最少困难标准(Least burdensome Standard) ”(FDA为提高审批的规范化和合理化,于1997年《食品、药品和化妆品法案》中提出的一项重要标准),加强医疗器械认证的关键信息,我们鼓励并帮助创新者提供高质量证据,以尽可能提高获得营销批准的效率。这些政策收效明显。2017年,FDA新批准的医疗器械数量达到95个,创2009年以来的最高纪录,是2009年获批数量的4倍多(如图3所示)。
2017:推进FDA监管程序改革
医疗科技日新月异的同时,也给监管带来了新的挑战。面对基因治疗、靶向药物、基于细胞的再生医学和数字化健康等高精尖科学领域的管理,传统的监管模式受到挑战。为了迎接这些新的挑战,我们以新思路审视并调整合适的监管模式,在产品评估过程中,既确保有利的技术进步,又能够一如既往地保护消费者权益。
新的改革方案允许某些诊断测试由获得资质的第三方审查,减轻药物测试开发者的负担、简化创新产品的监管评估流程。这种方案更适应新技术的高度迭代性,而其中的测试方法也需要常规修改以增加其精确度和临床效用。
2017年夏启动的健康数字创新试点计划(以下简称“计划”)旨在探索数字医疗器械的监管方法。快速发展的数字技术迅速演变为产品,是医疗应用程序等软件工具的特点。通过规范数字医疗技术并跟踪开发过程,可以保证数字健康工具的可靠生产。该计划为低中风险的医疗器械制造商明确了要求,在保证器械安全可靠的基础上更快地升级产品,使患者受益。
与该计划类似,再生医学综合规定和医疗设备3D打印制造商指导方案也根据新技术的独特属性,鼓励安全、有效地开发潜在的变革性产品。
监管程序改革并不限于上市前审查过程。为确保医疗产业安全,改革方案也出台了上市后监管的相关政策,为患者提供更安全的产品。做好上市后的市场监督,使患者获得更全面、科学的医疗决策指导信息,并能更有效地将产品推向市场。例如,在2017年秋启用的“药物不良反应”数据库中,患者和医疗保健人员可搜索药物和生物制品不良事件的报告,获得重要临床信息。这一举措大大增强了潜在风险药品的上市后监管力度,提前告知消费者药物的潜在问题,可有效预防无良诊所向患者提供如高风险干细胞产品等违法行为,保障民众安全。
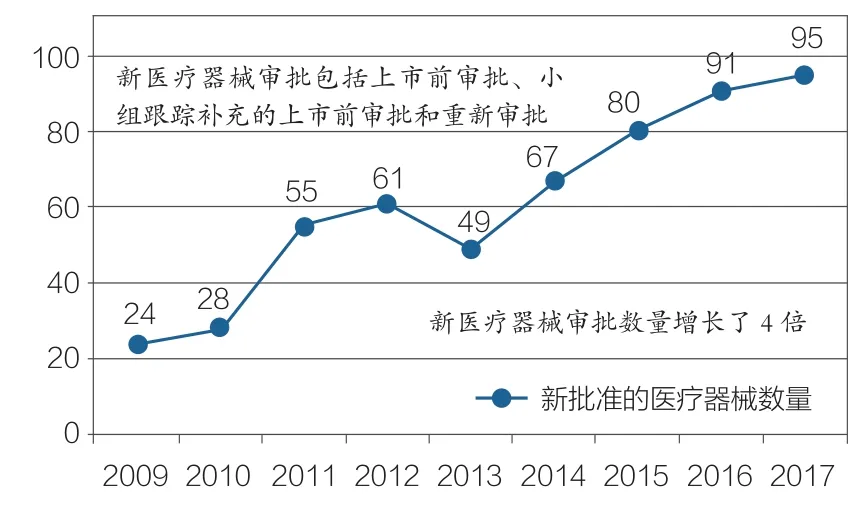
图3 2009-2017年FDA新型医疗器械审批情况
企业虚假声明草案(以下简称“草案”)则针对企业虚假发布其未经批准药品的治疗功效。草案根据药品的潜在风险调整顺势疗法药品审批标准, 提醒公众留意未经证实或测试的产品的潜在危害,如某些受污染的膳食补充剂。
FDA将继续推动大数据在监管和决策中的应用。
2017:促进药品市场良性竞争
虽然FDA无权来调节药品价格,但却有责任确保医药产业公平竞争的市场环境,抑制药价上涨,保障患者放心使用。
作为监管机构,FDA是药品公平竞争的基础力量。2017年6月推出的药物竞争行动计划(以下简称“计划”),旨在让患者获得更多价格优惠合理的药物。该计划优先审查仿制药物,防止公司延迟仿制药上市,清除了仿制药的审查积压,确保低成本药物能够以更实惠的价格让患者受益。
2017:维护消费者权益
除了医疗产业政策的改革创新,食品安全现代化法案(以下简称“法案”)的实施也使公众受益。该法案作为近70年来最全面的食品安全改革举措,旨在保护公众免受食源性疾病的威胁。2017年7月,FDA宣布提供3000万美元的资金帮助各地的食品生产者和农民等执行新的农产品安全要求。同时推出的“食品安全制定计划”软件可协助制造商制定食品安全计划,预防食源性污染,保护公众健康。
2017年,FDA举行了首次有患者参与的咨询委员会会议,加强与患者的接触,确保在监管决策中听到来自患者的声音。在重大疾病的治疗中,患者希望更早地进入实验性治疗。为加速罕见疾病药物开发,FDA发布了相关的指导意见草案,规定在罕见疾病药物的相同临床试验中,企业可合作测试多种药物产品以减少需要用安慰剂治疗的患者数量。改革还包括扩展患者的信息资源、扩展在线接入导航工具、简化批准患者接受调查治疗的审批过程。
2017:改善重要药物的管理
抗生素的耐药性仍然是一个重大的公共卫生挑战。解决这个问题需要采用“壹健康(One Health)”方法:在人类和动物医学中更负责任地使用抗生素。为了更好地管理食用动物的抗生素使用,FDA完成了一项开创性的行业指导方案(GFI#213),禁止在食用动物中使用医疗抗生素,其他治疗需要在兽医监督下进行。
指导方案中涉及292个批准的药物申请,其中84个药物申请被撤回,93个申请从非处方药(OTC)转化为处方药口服剂型产品,另外115个申请从OTC转化为兽用饲料指令的产品。该方案对引导合理使用抗生素治疗动物疾病有重要作用。2018年将出台更多措施来巩固这些成就,并改善医学上重要抗生素的管理。
2018展望
新的一年,我们有望在更多领域采取措施推进有益创新,确保药物生产过程高效、安全,实施保护和促进公众健康的切实解决方案。在议程上,FDA也将优先推进生物仿制药改革,推进非处方药产品现代化,更好地向女性警示健康问题和风险因素。
没有人能够独自创新。医学、生物技术、食品科学和整个公共卫生的进步,只有合作才能实现。FDA的独特地位可以将整个行业(患者、产业、学者、供应商和其他政府机构)的相关方凝聚在一起,确保生化医疗领域的创新切实、高效地转化为惠及公众的应用成果,这将是我们在未来一年追求并推动的重要目标。
猜你喜欢
中老年保健(2022年1期)2022-08-17
中老年保健(2021年9期)2021-08-24
中老年保健(2021年12期)2021-08-24
劳动保护(2019年7期)2019-08-27
中国卫生(2016年1期)2016-11-12
中国卫生(2016年1期)2016-11-12
中国卫生(2016年1期)2016-01-24
中国卫生(2015年7期)2015-11-08
中国卫生(2014年6期)2014-11-10
中国卫生质量管理(2014年5期)2014-02-28
