
柴油馏分碳数分布的预测研究
2018-12-11 07:47任小甜褚小立田松柏
石油炼制与化工 2018年12期
任小甜,褚小立,田松柏
(中国石化石油化工科学研究院,北京 100083)
对于柴油来说,其沸程、密度等各项物性以及十六烷值、润滑性、低温流动性等使用性能都与其分子组成有关[1],不同来源(不同产地的原油和不同的加工工艺)的柴油分子组成差别较大,其从根本上决定了柴油的各项性质及其使用性能。传统的表征方法只能得到柴油馏分的烃类组成,为了满足目前清洁、优质柴油的生产需求,需要从分子水平上深入认识柴油的详细组成,探究其各项物性及使用性能和分子组成的关系。目前,基于软电离技术的气相色谱飞行时间质谱(GC-TOF MS)是一种快速有效的柴油分子组成的表征方法[2-3]。但该仪器平台价格昂贵,分析过程比较复杂,需要专业的技术人员进行操作,不能满足工业生产中快速实时分析的要求。而柴油的基本物性由常规分析手段就可以得到,大多数炼油厂都具备这样的分析条件;另外,由柴油馏分的基本物性数据,尤其是近红外光谱数据又可以准确计算其烃类组成信息[4]。如果能够利用这些常规物性和组成数据快速预测柴油馏分的详细碳数分布信息,将对石油的分子水平炼制产生重要影响。
现有文献已经报道了一些基于常规物性计算来预测柴油馏分的分子组成包括碳数分布组成的方法[5-6],其基本研究思路是预先设定一个虚拟分子库,确定每个虚拟分子的各项物性数据,再利用混合规则来计算柴油馏分的宏观物性,最后通过优化各项宏观物性的损失函数来确定各分子的含量,以这些虚拟分子的组成来表示柴油。这种方法存在很大的局限性,首先,虚拟分子的设定不一定准确,而且需要确定大量单体分子的各项物性数据,许多物性存在缺失的情况,准确度也不能保证;另一方面,从优化计算的角度来讲,虚拟分子的数量远大于宏观物性的数目,这样会导致计算结果不唯一,即使引入人为简化的分布函数减少变量个数,其本质也是一个非凸优化的过程,求解难度很大,准确度也不够高。
本研究从机器学习的角度出发建模,以一定数量的柴油样本为基础,确定其标准的物性数据和烃类组成数据作为特征,以其详细的碳数分布组成数据作为输出,结合最近邻回归算法(KNR)和过采样技术建立回归模型,实现由柴油馏分的物性数据和烃类组成数据快速预测其详细碳数分布组成信息。
1 算法原理
1.1 KNR算法
最近邻算法(KNN)是一种有监督的机器学习算法,不同于其它算法的是,其属于消极学习,即模型不需要训练,无需估计参数,根据输出目标种类的不同又分为KNR和最近邻分类算法(KNC)。该算法的基本思想是距离相近的样本具有相似的属性,预测时先分别计算待测样本到每个训练集样本在特征空间中的欧式距离,找出距离最近的k个样本,再利用最邻近的这k个样本进行决策,对于分类问题,k个样品中哪种类别的训练样本占多数,待测样本就属于这一类,对于回归问题,则将k个样本属性的平均值作为待测样本的属性。对于柴油馏分,假设各项物性数据和烃类组成相似的样本,其本质上就是相似的,反映到分子组成上就是其碳数分布组成信息都是类似的。所以,本研究选择KNR算法建模,即先根据物性数据和烃类组成找到与待测样本距离最近的k个训练样本,将这几个样本的碳数分布组成数据按照距离的不同进行加权线性加和,即同时得到待测样本的包含300多项集总含量的碳数分布信息。其中,引入高斯函数来计算不同距离样本的权重ω,计算式如下:
(1)
(2)
式中:k为最近样本的个数,Si表示第i个最近样本的高斯函数值;ωi为第i个最近样本对应的权重;di为待测样本和第i个最近样本的欧式距离;σ为高斯函数的参数,本研究取值为10。待测样本的碳数分布组成数据的预测计算式如下:
(3)
式中:y为待测样本的碳数分布组成数据,yi为k个最近样本中第i个样本的碳数分布组成数据。
1.2 过采样技术
对于KNR算法,库中的训练样本越多,越能找到与待测样本距离更近的样本,即碳数分布组成信息相似的样本,从而保证预测结果的准确性。而在实际应用场景中,收集大量组成数据和物性数据都完备的样本是比较困难的,尤其是对于分析成本较高的性质,如碳数分布组成信息等。另外,对于不同待测样本,尤其是不同种类的样本,很难保证在库中能找到距离足够近的样本来满足KNR的计算。所以,本研究利用过采样技术来解决训练样本不足的问题。该算法的基本思想是,先从库中找到与待测样本距离最近的几个样本,然后将这几个近邻样本按照任意质量比进行混合,这样可以得到大量的虚拟样本,同时虚拟样本的物性数据和组成数据也可以由线性加和计算得到,其计算式如下:
(4)

利用过采样技术,在每个待测样本的周围都生成大量的、物性和组成数据完备的虚拟样本,这样就可以保证KNR算法能准确地找到最相似的样本,进而保证预测结果的准确性。需要注意的是,要利用过采样技术生成虚拟样本,库中各样本的各项属性都必须满足线性加和的规则,或者经过数学转换后符合线性加和规则。具体到柴油馏分中,就是要选择符合线性加和的物性数据作为输入特征,烃类组成数据和碳数分布组成信息自然是满足线性加和条件的。
2 实 验
2.1 测定样品数据
收集直馏柴油样品78个,这些样品从不同产地的原油分馏而得,具有一定的代表性。用标准方法测定其硫含量、氮含量、酸值,这3项物性能满足线性加和条件。用气相色谱-质谱联用仪分析柴油样品的烃类组成信息(SHT 0606),用带场电离源(FI)的GC-TOF MS测定样品的详细碳数分布信息。
2.2 数据预处理
以柴油样本的3个物性数据、11个烃类(分别为链烷烃、单环环烷烃、双环环烷烃、三环环烷烃、烷基苯、茚满四氢萘、茚类、萘类、苊类、苊烯类和三环芳烃)的组成信息作为模型的输入特征X。对于碳数分布信息,根据标准方法中的模板,用一个由碳数分布范围在7~30的13种烃类同系物的族组成的矩阵表示,将该碳数分布矩阵按照不同的列依次展开,共计312个碳数项,构成模型的输出值Y,由此确定模型的数据库。
对于KNR算法,其中会涉及到样本间距离的计算,而柴油样品的物性数据和烃类组成数据的量纲不同,取值范围也有很大的差异,所以要对样本的输入特征X进行标准化预处理,消除量纲的影响,进一步保证预测结果的准确性。
2.3 建立预测模型
上述库中的78个样本作为训练集。模型计算的流程如下:取一个待测样本,首先将其输入特征X进行标准化预处理,再计算其与库中各样本的欧式距离,找出最近的6个样本,利用过采样技术生成5 000个虚拟样本,将待测样本周围的特征空间密集化;然后通过KNR算法,从生成的虚拟样本中找出最近邻的k个样本,然后通过线性加权求和计算出碳数分布组成的预测值,并与实验值进行比对。
3 结果与讨论
3.1 相似度分析
本研究的模型以柴油的3项物性和11个烃类组成数据作为输入特征,这些特征能否充分反映出样本的本质区别是模型准确预测的先决条件,即通过这些特征从库中找出两个相似样本,在碳数分布组成上也必须保持基本一致的相似度。为此,本研究考察了样本在特征和碳数分布组成上的相似度,随机抽取一个柴油样本CY656,从库中找出与其欧氏距离最近的3个样本(CY540,CY621,CY624),分别计算这3个近邻样本和待测样本CY656在特征和碳数分布组成上的相似度(sd1、sd2),计算结果如表1所示。其中相似度的计算式如下:
(5)
式中:sd表示两个样本之间的相似度;d表示样本之间的欧式距离;m表示特征的维度。

表1 近邻样本的相似度比较
计算结果表明,待测样本CY656与近邻样本CY540,CY621,CY624在特征和碳数分布组成上的相似度基本一致,根据直馏柴油的14项特征可以从库中找出与待测样本碳数分布组成基本相似的样本,进而可以用这些近邻样本拟合计算待测样本的碳数分布组成,因此本研究的预测模型能保证足够的准确度。
3.2 模型的超参数
KNR模型中一般有一个重要的超参数需要优化,即最近邻样本数目k,若k值太小,模型的泛化性能则很差,很可能在库中找不出k个最近邻样本;假如k值太大,则可能找出不是最相似的样本,模型的准确性又下降。本研究利用交叉验证的方法来确定模型中k的最优取值。在库中任意抽取5个样本,利用过采样技术在这5个样本之间生成5 000个虚拟样本,然后用这些样本构建KNR的10折交叉验证模型。首先将样本平均分为10份,依次取出1份作为测试集,剩下的9份作为训练集,接着利用KNR算法分别对每1份测试集的碳数分布组成进行计算,并计算这些训练集样本的平均标准偏差(RMSECV)。选定不同的k值(1~15),依次进行上述的计算,根据RMSECV的取值最小确定模型的最佳k值。超参数k的交叉验证计算的结果如图1所示。由图1可知,当k=5时,RMSECV的取值最小,说明模型的最佳k值为5。
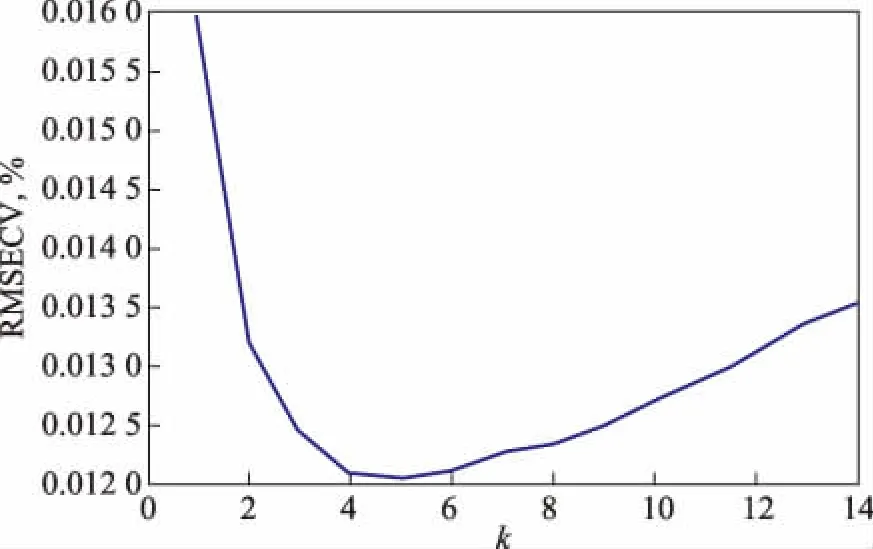
图1 k值的10折交叉验证计算
3.3 待测样本的预测分析
新取一个待测样本,用标准方法测定其硫含量、氮含量、酸值以及11项烃类组成数据,将这14个输入特征X进行标准化预处理,然后代入上述的预测模型进行碳数分布组成的计算。将14个输入特征进行主成分分析,选取前两个主成分的得分(PC1、PC2)进行作图,样本在特征空间中的分布如图2所示,其中红色的点为待测样本,黑色的点为与待测样本相似的6个库中真实样本,蓝色的点表示由这6个近邻样本生成的5 000个虚拟样本。由图2可知,通过过采样技术,可以将待测样本周围的空间有效地密集化,进而满足算法的要求,保证预测模型的准确性。

图2 样本在特征空间中的分布
石油中的化合物类型通常用缺氢数Z加上杂原子来表示,分子中每增加1个双键或者1个环,Z值就减少2,如苯并噻吩类化合物可由-10S表示。柴油的碳数分布组成数据中一般包括13种分子类型,分别为链烷烃、单环环烷烃、双环环烷烃、三环环烷烃、烷基苯、茚满四氢萘、茚类、苯并噻吩、萘类、苊类、苊烯类、二苯并噻吩和三环芳烃,对应的Z值分别为+2,0,-2,-4,-6,-8,-10,-10S,-12,-14,-16,-16S,-18。待测样本的饱和烃、单环芳烃、双环芳烃和三环芳烃的碳数分布对比如图3所示,其中折线为模型的预测值,散点为实测值。从图3可以看出,饱和烃和双环及三环芳烃的碳数分布的预测偏差都比较小,预测值和实测值都比较吻合,单环芳烃在低碳数处有一定的预测偏差,总的来说,本研究所建立模型的预测精度较高。图4为整个碳数分布矩阵预测值和实测值的气泡图对比。从图4可以看出,所有气泡基本都能完全契合,只是在少数含量很低的高碳数组分处出现了一定的偏差。上述结果表明,本研究的模型预测精度高,能得到和标准分析方法基本一致的结果,具有一定的应用价值。此外,在预测时发现,当待测样本处于界外,即在库中找出的近邻样本相距都较远时,就会出现预测偏差增大的情况,在样本的空间分布图上也会显示出虚拟样本不能包围覆盖待测样本,所以必须保证库中有大量样本,在后续的模型维护中,主要的工作就是增加各类型的柴油馏分样本。
这种模型不需要训练,没有参数估计,方便部署,计算速度快,准确度高,可以一次性计算出多达312项的碳数分布信息,另一方面,模型的维护也比较容易,面对一个新的应用环境时,不需要再进行训练,直接在库中增加样本即可。同时,对于不同种类的柴油馏分,也不需要分别单独建模,只要保证库中有足够的各类型样本,再结合过采样技术同步在待测样本周围生成虚拟样本,就可以给出比较准确的预测。
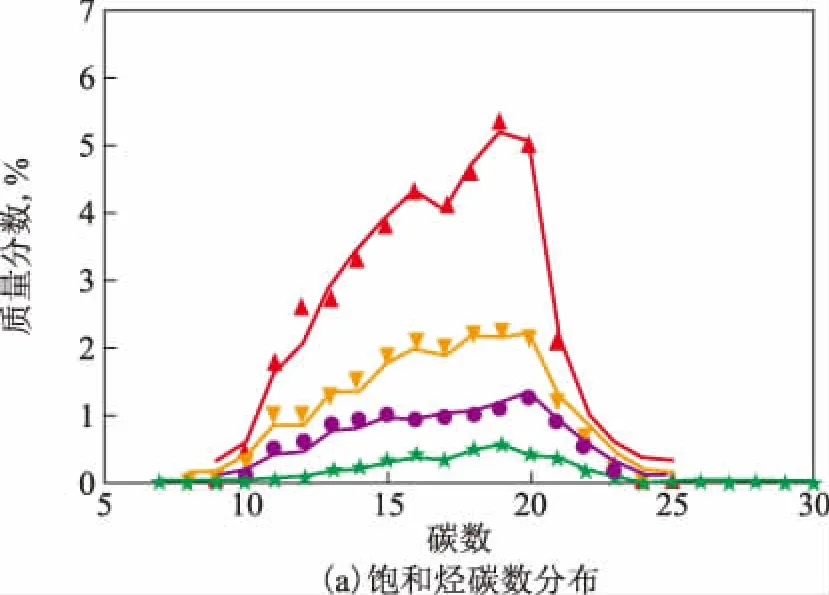
模型预测值: —链烷烃; —单环烷烃; —双环烷烃; —三环烷烃。实测值: ▲—链烷烃; 单环烷烃; ●—双环烷烃; ★—三环烷烃

模型预测值: —烷基苯; —茚满或四氢萘; —茚类; —苯并噻吩。实测值: ▲—烷基苯; 茚满或四氢萘; ●—茚类; ★—苯并噻吩

模型预测值: —萘类; —苊类; —苊烯类; —二苯并噻吩; —三环芳烃。

图4 碳数分布的预测值和实验值气泡图
4 结 论
(1)结合KNR算法和过采样技术提出了一种由物性数据和烃类组成数据快速预测柴油馏分中碳数分布组成的方法。
(2)利用上述算法,对直馏柴油进行了建模研究,以柴油的硫含量、氮含量、酸值以及11项烃类组成(分别为链烷烃、单环环烷烃、双环环烷烃、三环环烷烃、烷基苯、茚满四氢萘、茚类、萘类、苊类、苊烯类和三环芳烃)信息作为模型的输入特征进行模型计算,结果表明该方法预测精度高,能一次性快
速地计算出直馏柴油中312项碳数分布组成信息,同时模型维护简单,通过增加库中的样本就可以扩大模型的适用范围,具有一定的实用价值。
猜你喜欢
石油学报(石油加工)(2022年4期)2022-07-19
石油化工(2022年3期)2022-03-29
能源工程(2022年1期)2022-03-29
韩国语教学与研究(2021年1期)2021-07-29
煤气与热力(2021年6期)2021-07-28
环境科学研究(2020年2期)2020-03-03
石油化工应用(2019年7期)2019-08-13
天然气化工—C1化学与化工(2018年5期)2018-11-15
科技创新与应用(2018年4期)2018-01-31
中学生数理化·高二版(2017年3期)2017-07-07
