
Dissociation and physical properties of methyl iodide in external electric field*
2018-12-11 03:18ZHANGXiangyunLIUYuzhuMAXinyuQINChaochao
中国科学院大学学报 2018年6期
ZHANG Xiangyun, LIU Yuzhu ,2†, MA Xinyu, QIN Chaochao
(1 Jiangsu key Laboratory for Optoelectronic Detection of Atmosphere and Ocean, Nanjing University of Information Science & Technology, Nanjing 210044, China; 2 Jiangsu Collaborative Innovation Center on Atmospheric Environment and Equipment Technology (CICAEET), Nanjing 210044, China; 3 College of Physics and Materials Science, Henan Normal University, Xinxiang 453007, Henan, China)
Abstract As a kind of toxic methylation reagent and disinfectant, methyl iodide (CH3I) is widely used. It is important to study the basic physical properties of methyl iodide and to use effective measures to degrade it. The ground states of methyl iodide in different electric fields from 0 to 0.04 a.u.(atomic unit) are optimized using the B3LYP calculation with the LANL2DZ basis set. Optimized parameters, total energies, bond lengths, dipole moments, the highest occupied molecular orbital energies, the lowest unoccupied molecular orbital energies, energy gaps, infrared spectra, and dissociation potential energy surface (PES) are obtained. The obtained results show that when the external electric field gradually increases from 0 to 0.04 a.u. along the molecular axis Z (the C-I bond direction), the total energy decreases while the dipole moment increases. The C-I and C-H bond lengths increase gradually. The energy gap first increases and then decreases with the external electric field. Further studies show that when the external electric field increases from 0 to 0.04 a.u., the dissociation PES along the C-I bond becomes unbound with the potential energy barrier disappearing. The external electric field of 0.04 a.u. is sufficient to induce the degradation of methyl iodide with the C-I bond breaking. This work provides an important support for the degradation of methyl iodide in the external electric field.
Keywords methyl iodide; external electric field; degradation; infrared spectrum
Methyl iodide is used primarily in the production of pharmaceutical intermediates and the organic synthesis for methylation reagents. It was considered as an alternative to soil disinfectants in the 1998 and 2002 Methyl Bromide Technical Options Committees Assessment. Methyl iodide is also widely used as a soil disinfectant to replace methyl bromide that severely destroyed the environment (prohibited by the Montreal Protocol Copenhagen Amendment). Methanol is toxic, corrosive, and carcinogenic, and methyl iodide naturally decomposes more slowly. At the same time, methyl iodide is widely used as a soil disinfectant but the possibility of entering the groundwater is very high. Inhalation of a large amount of methyl iodide inhibits the central nervous system and exerts strong stimulating effects on the skin and mucous membranes. Methyl iodine is easily decomposed by heat and produces toxic iodide fumes. It can be absorbed through the respiratory tract, skin, and digestive tract and causes poisoning. Domestic and foreign reports described how CH3I harmed people and animals and some poisoning incidents caused by CH3I[1,2].
In recent years, the degradation kinetics of halogenated compounds containing CH3I has received unprecedented attention[3-7]. Molecules in the external electric field will have a series of physical and chemical changes. The characteristic of molecules in external electric field has become an important method for studying molecular properties. This method has been successfully applied to a number of fields to study the properties of molecules[8-12]. Applying a strong electric field to a molecule to break off the chemical bond is an effective method for the degradation of the molecule. However, there is no reported study on the dissociation of the CH3I molecule in the external electric field.
The molecular total energy, molecular dipole moment, molecular energy gap, molecular spectrum, and dissociation properties of the CH3I molecule in external electric field (0~0.04 a.u. (atomic unit)) were studied using density functional theory at the B3LYP/LANL2DZ level. The Gaussian 09[13]software was used. The result provides important theoretical support for the degradation of CH3I.
1 Theoretical method
The Hamiltonian of the radiation process is given as
H=H0+Hint.
(1)
WhereH0is the complete molecular Hamiltonian,Hintis the interaction Hamiltonian between the electron field and the molecule. Under the dipole approximation,Hintis given in a.u. as
Hint=-μ·F
(2)
whereFis the radiation field,μis the dipole moment, and 1 a.u.(atomic unit)=5.142 25×1011V/m.
Based on the model proposed by Grozema et al.[14-15], excitation energy under the action of fieldEexc, electric field strengthFwith the variation amounts Δμ, Δαof the electric dipole moment and the polarization rate satisfies the relational expression
Eexc(F)=Eexc(0)-Δμ·F-1/2·Δα·F2.
(3)
Based on the geometric structure of the CH3I molecule without electric field, we use the B3LYP/LANL2DZ method of density functional theory to optimize the ground state structure and study the molecular total energy, molecular dipole moment, molecular energy gap, molecular spectrum, and dissociation properties of the CH3I molecule.
2 Results and discussion
2.1 Molecular stable structure without external electric field
Theoretical calculations show that the CH3I molecule has the C3 Vsymmetry. In the present work, we optimize the structure of CH3I with different methods. The experimental data[16]and optimization data are listed in Table 1. By comparing the calculated data with the experimental data, we can see that the structure parameters(the bond lengths and the bond angle) calculated at the B3LYP/LANL2DZ leved are the closest to the experimental data. Therefore, the dissociation and spectral characteristics of CH3I in the external electric field are calculated at the B3LYP/LANL2DZ level. The calculated stable structure is shown in Fig. 1. TheX-axis,Y-axis, andZ-axis are delined according to the Dicker coordinate system, and theZ-axis is along the the C-I bond.
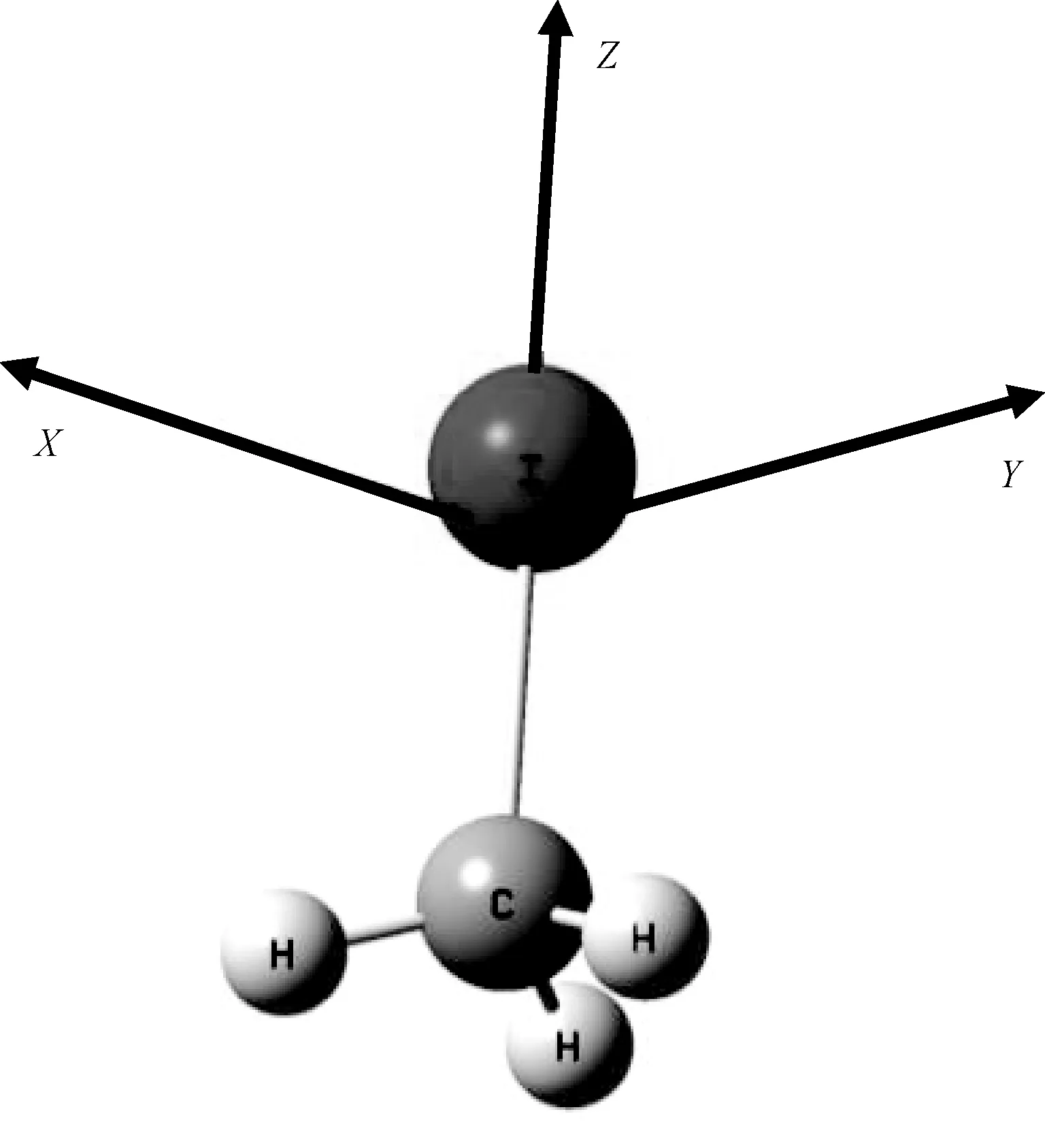
Fig.1 Optimized geometry of the ground-state CH3I

Table 1 Optimized parameters of the structure of CH3I at different levels, together with the experimental data
2.2 Effect of external electric fields on bond length and total energy
The CH3I molecule was optimized and calculated at the B3LYP/LANL2DZ level when different electric fields (0~0.04 a.u.) were applied in theZ-axis (along the C—I bond). The stable structures of methyl iodide at different field strengths were obtained. The calculated bond length, and dipole moments of the CH3I molecule are given in Table 2. It can be seen in Table 2 that the total energy decreases gradually with the increase of the external electric field (0~0.04 a.u.). The C—I and C—H bond lengths increase gradually with the increase of the electric field (0~0.04 a.u.) in theZ-axis. The results show that the chemical bond length increases gradually with the external electric field and the modecule becomes more and more prone to dissociation. With the increase of the external field from 0 to 0.04 a.u. along theZ-axis, the dipole moment increases.
2.3 Effect of external electric fields on the molecular orbital energies
It is well known that many properties of a molecule are determined by the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The HOMO and LUMO energies of the CH3I molecule are obtained at the B3LYP /LANL2DZ level. The HOMO energyEH, the LUMO energyEL, and the energy gapEGare shown in Table 3.EGis given as
EG=(EL-EH)×27.2 eV.
(4)
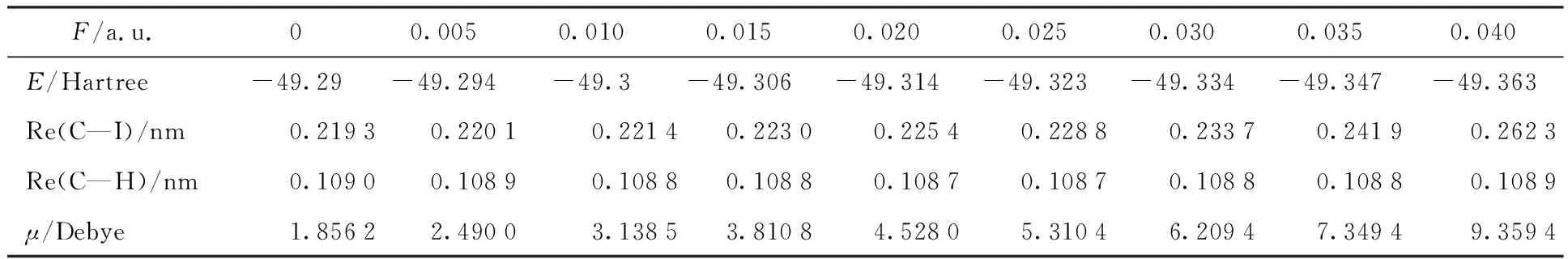
Table 2 Calculated molecular total energies, bond distances (C—I and C—H),and dipole moments of CH3I in different external electric fields
It can be seen in Fig. 2 that with the increase of electric field the HOMO -LUMO energy gapEGgradually increases first and then decreases sharply. It is shown that at the beginning the methyl iodide molecule is hard to be excited to the excited state. When the electric field strength is greater than 0.02 a.u., molecule are getting more and more easily excited. It is also shown that the ability of methyl iodide to participate in chemical reactions weaks and then becomes strong.

Table 3 Calculated LUMO energy EL, HOMO energy EH, and HOMO-LUMO energy gap EG for CH3I in different external fields
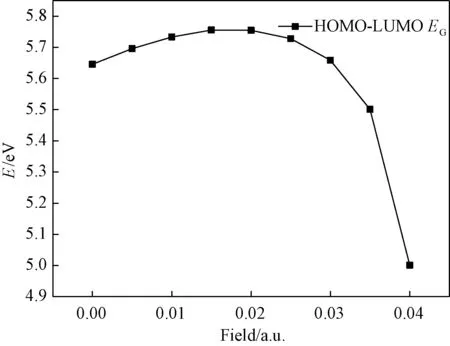
Fig.2 Variations in the HOMO-LUMO energy gap of CH3I in external fields
2.4 Effect of external electric fields on IR spectra
The IR spectra of CH3I were calculated at the B3LYP/LANL 2DZ level. The calculated IR spectra mainly correspond to six kinds of vibration peaks, as shown in Table 4. Frequency of 481.67 cm-1is attributed to the CI str vibration, frequency of 890.77 cm-1is attributed to the CH3rock vibration, frequency of 1 282.34 cm-1is attributed to the CH3s-deform vibration, frequency of 1 431.46 cm-1is attributed to the CH3s-str vibration, frequency of 2 981.21 cm-1is attributed to the CH3s-str vibration, and frequency of 3 110.33 cm-1is attributed to the CH3d-str vibration. As shown in Table 4, the calculated symmetries and vibration frequencies are in agreement with the experimental results[17], with errors of no more than 10%.

Table 4 Calculated IR parameters for CH3I including vibrational frequencies and symmetries,together with the experimental data
In Table 4, str denotes the stretching vibration, and deform denotes the bending vibration. The IR spectra of the CH3I molecule under different external electric fields were calculated by applying different intensity of electric fields (0~0.04 a.u.) in the direction of the C—I bond. The external electric field has significant influences on the IR spectra of the CH3I molecule.
2.5 Effect of external electric field on molecular potential energy curve
Without external electric field, we carried out energy scanning for CH3I along the C—I bond direction at the B3LYP /LANL2DZ level to obtain the potential energy curve. The same method was used to calculate the potential energy curves along the C—I bond direction with different electric fields along theZ-axis (the C—I bond direction). The potential energy curves without and with the field are plotted in Fig. 3. It is shown that the potential energy curves of the CH3I molecule are gradually repulsive in the external electric fields (0~0.04 a.u.). When the external electric field is 0.04 a.u., the potential energy curve of CH3I changes from “bound” to “repulsive”. The CH3I molecule will be degraded at the strength of 0.04 a.u.. The electrochemical degradation of the CH3I molecul will be important for the protection of the environment.

Fig.3 Variation of the potential energy curve along the C—I bond of CH3I in external field
3 Conclusions
In the present work, the dissociation characteristics and spectral characteristics of CH3I in external electric field were calculated using the principle of density functional theory. It is found that the results obtained at the B3LYP/LANL2DZ level are the closest to the experimental data. Therefore, the B3LYP/LANL2DZ method was used to calculate the configuration, IR spectrum, and dissociation potential energy curve of CH3I in different electric fields. When a series of external electric fields are applied along theZ-axis (the C—I bond), the energy of the molecular system gradually decreases. The dipole moment decreases, indicating that the polarity is constantly increasing. HOMO -LUMO energy gapEGincreases first and then decreases sharply, indicating that at the beginning, the methyl iodide molecule is hard to be excited into the excited state. When the electric field strength is greater than 0.02 a.u., the excitation of the molecule to the excited state will become more and more simple. It is also shown that the ability of methyl iodide to participate in chemical reactions weakens and then becomes strong. The C—I and C—H bond lengths monotonously increase. The external electric field also has effects on the positions and intensities of the IR spectra of the CH3I molecule.The potential energy curve of the CH3I molecule along the C—I bond changes gradually from “bound” to “repulsive”. It is found that the electric field of 0.04 a.u. is sufficient to cause the C—I bond breakage of CH3I, which provides an important scientific basis for the degradation of CH3I.
