
高效液相色谱-串联飞行时间质谱高通量筛查生鲜乳中兽药残留
2018-11-28 06:52田怀香何玉洁周兴鑫郑小平何亚斌于海燕
食品科学 2018年22期
田怀香,何玉洁,周兴鑫,郑小平,何亚斌,于海燕,陈 臣,*
(1.上海应用技术大学香料香精技术与工程学院,上海 201418;2.上海必诺检测技术服务有限公司,上海 200436)
乳及乳制品因其高营养等特点在世界范围内的发展速度有目共睹,是当今最具发展潜力和前景的食品之一。作为乳及乳制品的源头,生鲜乳的安全保障是一个关键环节。其中,生鲜乳中兽药残留问题备受关注[1]。针对兽药残留,我国农业部235号公告[2]中作出了明确的限量要求,实际检测中需要严格遵守。但违规使用兽药的情况仍然非常严重[3]。
常用的兽药残留检测方法包括电泳法[4]、气相色谱法[5-6]、液相色谱法[7-8]、色谱-质谱联用法[9-14]等。气相色谱法检测涉及衍生操作,适用性窄;液相色谱法分离效率较低,专属性不强;液相色谱-串联质谱联用法作为使用广泛的兽药残留检测方法,虽然灵敏度高,在定性定量方面具有较强优势[15-17];但是在大通量筛查时,其检测兽药化合物的数量有限,通常为几十种[18-19]。
高效液相色谱-串联飞行时间质谱(high performance liquid chromatography tandem time-of-flight mass spectrometry,HPLC-TOF-MS/MS)法相比低分辨率的液相色谱-质谱而言,缩短分析时间的同时,还大大提高了分析的化合物数目[20-21]。一次进样分析中,TOF可进行一次快速全扫描,获得所有母离子信息;此外,结合信息相关分析模式(information dependent acquisition,IDA),对达到一定强度的子离子进行二级碎片的确证,使定性更加精确。Martin等[22]利用HPLC/Q-TOF-MS方法对废水和河水中阿托伐他汀、氟伐他汀等6 种抑制素的提取方法进行了优化,并分析对比3 种提取方法的仪器方法检出限,分别为固相萃取(0.04~11.2 ng/L)、分散液液萃取(0.10~17.0 ng/L)、搅拌棒吸附萃取(0.52~2.00 ng/L),具有较高的灵敏度,Gu Junfei等[23]用此法结合固相萃取分析检测了老鼠血清中4 种马兜铃酸,在日内和日间出峰保留时间的相对标准偏差为1.1%~1.5%,具有较好的稳定性。Song Haojing等[24]用HPLC/Q-TOF-MS/MS第一次检测岩白菜素在老鼠体内的代谢产物,检测样主要为老鼠肉、胆汁、尿液和粪便,检测结果通过总离子色谱图和二级离子质谱碎片图光谱信息为目标物及代谢碎片化合物进行定性分析等。
鉴于国内对于高通量兽药筛查的研究较少,本研究主要利用HPLC-TOF-MS/MS方法对生鲜乳中可能存在残留风险的兽药化合物进行筛查。这些兽药涵盖了β-内酰胺类、β-兴奋剂、阿维菌素类、苯并咪唑类、大环内酯类、非甾体类消炎药、磺胺类、抗球虫剂、喹诺酮类、类固醇类、林可酰胺类、氯霉素类、四环素类、糖皮质类、硝基咪唑类、镇静剂和其他共17 类兽药化合物,为实际生鲜乳中兽药残留的筛查提供了一个简单且快速的检测方法,大大提高了筛查效率。
1 材料与方法
1.1 材料与试剂
本实验所用生鲜乳均来自上海各牧场,其中经检测不含目标待测化合物的生鲜乳即为空白样品。
β-内酰胺类、β-兴奋剂、阿维菌素类、苯并咪唑类、大环内酯类、非甾体类消炎药、磺胺类、抗球虫剂、喹诺酮类、类固醇类、林可酰胺类、氯霉素类、四环素类、糖皮质类、硝基咪唑类、镇静剂和其他共17 类兽药化合物,共174 种兽药化合物标准品 美国Sigma Aldrich公司。所有标准品用甲醇或水配制成一定浓度的标准储备液,于-20 ℃储存。中间液用甲醇配制,质量浓度为10 µg/mL,于-20 ℃储存;临用时,配制成质量浓度为200 ng/mL的混合标准工作液,于-20 ℃储存。
甲醇、乙腈、乙酸乙酯、二氯甲烷(均为色谱纯)美国Thermo公司;甲酸、氯化钠、无水硫酸钠(均为色谱纯) 德国Merck公司;去离子水由Milli-Q超纯水系统生产。
1.2 仪器与设备
Triple TOF 5600 MS仪(配有电喷雾离子源) 美国AB SCIEX公司;ACQUITY HPLC仪、Oasis PriME萃取柱(60 mg/3 mL) 美国Waters公司;Cleanert PEP-Plus(60 mg/3 mL)萃取柱 美国Agela公司;SORVALL ST 16R离心机 美国Thermo公司;旋转蒸发仪德国IKA公司;氮气浓缩仪 上海安谱实验科技股份有限公司;Milli-Q超纯水制造仪 美国Millipore公司。
1.3 方法
1.3.1 样品前处理
精确称取生鲜乳样2 g于50 mL离心管中,加入混合标准工作溶液500 µL,振荡,静置10 min。加入1 g氯化钠和1 g无水硫酸钠,振荡。加入10 mL的乙腈,涡旋振荡处理5 min,于10 000 r/min离心5 min,转移上清液至另一50 mL离心管中。再加入10 mL的乙腈溶液重新提取一次,同样条件离心后上清液合并。再加入10 mL乙酸乙酯提取一次,同样离心条件离心后合并上清液,于50 ℃旋转蒸发至近干,1 mL甲醇复溶。于-20 ℃冷冻2 h以上,于10 000 r/min离心5 min,过0.22 µm有机滤膜,上机检测。
既往研究表明Ki67表达率以及人表皮生长因子受体2(HER-2)与nSLN转移与否无相关性[11]。Ki67表达率≤30%时,nSLN转移率为45%(14/31);而Ki67表达率>30%时,nSLN转移率为42%(14/33)。单因素分析结果显示,组间差异P值=0.825,无显著统计学差异。HER-2为阳性时,nSLN转移率为50%(8/16);而HER-2结果为阴性时,nSLN转移率为42%(20/48)。单因素分析结果显示,组间差异P值=0.561,无显著统计学差异。
1.3.2 色谱条件
色谱柱:Poroshell 120 EC-C18(3.0 mm×150 mm,2.7 µm);流动相:0.1%甲酸溶液和甲醇;流速:350 µL/min;柱温:35 ℃;进样量:5 µL;线性梯度洗脱程序见表1。

表1 液相色谱梯度洗脱程序Table 1 Gradient elution program
1.3.3 MS条件
离子源:电喷雾离子源(electron spray ionization,ESI);扫描模式:正离子扫描(ESI+);检测方式:全离子扫描;扫描范围m/z 100~1 000;电喷雾电压:5 500 V;雾化气流量和辅助气流量均为55 L/h;气帘气流速35 L/h;离子源温度600 ℃。去簇电压80 V;碰撞能量10 V;离子累积加速时间0.25 s。
在上述MS条件下,增加信息相关扫描,监测响应值超过1 000的化合物,得到其子离子信息,扫描范围m/z 50~1 000,去簇电压60 V;碰撞能量(35±15)V;扩展碰撞能量15 V;离子累积加速时间0.1 s。
一级全扫描获得的高精度相对分子质量数据用于筛选及定性分析,IDA全扫描获得的二级碎片离子信息用于加强定性确证。
1.4 数据及图像处理
采用仪器Peak View软件对筛查谱图进行定性分析,Multi Quant软件对方法验证中回收率等指标进行分析处理;采用Excel软件分析精密度;采用Origin 9软件对图像进行调整处理。
2 结果与分析
2.1 标准筛查谱库的建立结果
用200 ng/mL的混合标准溶液,建立174 种目标兽药化合物的筛查谱库,相比于国内现有的兽药残留检测报道[25-27],种类、数量大大增加。其总离子流色谱图不能清晰地反应各化合物的具体信息,图1为苯并咪唑类物质的提取离子色谱图和二级碎片谱库比对图,174 种目标兽药化合物的质谱相关信息和保留时间见表2。
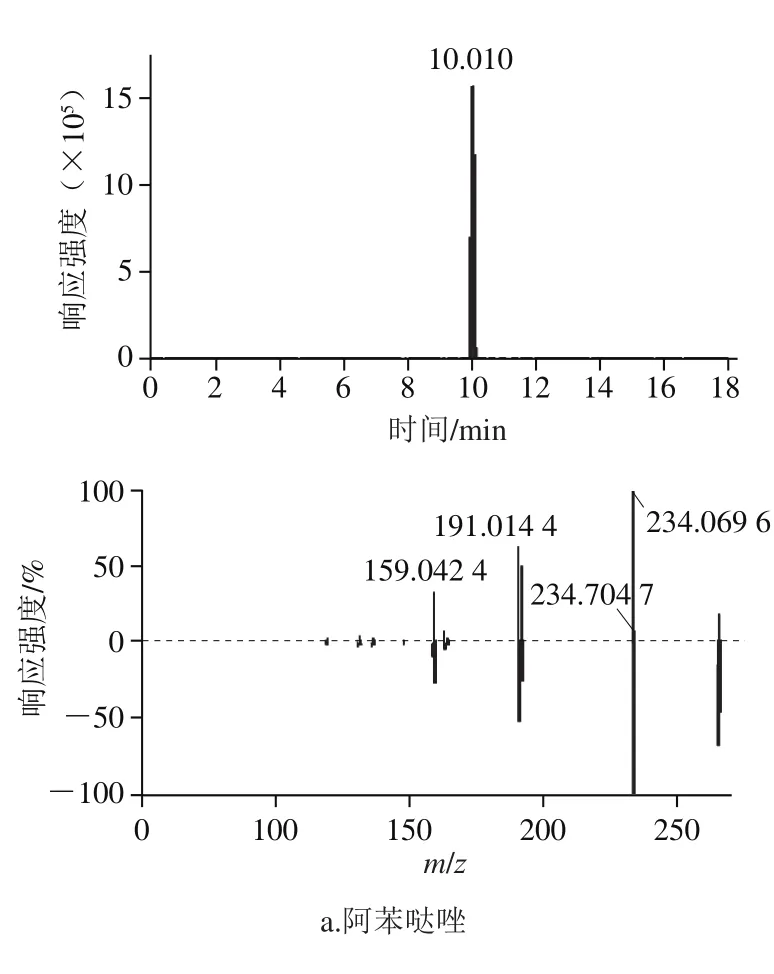


图1 9 种苯并咪唑的选择离子色谱图和二级离子色谱图信息Fig. 1 Extracted ion chromatograms and secondary ion chromatogram information of nine benzimidazoles
2.2 净化方法的优化结果
本实验比较过柱净化和冷冻净化对于目标分析兽药化合物的回收情况。所选用净化小柱为Oasis PRiME柱和Cleanert PEP-Plus柱。过柱净化步骤:3 mL甲醇、3 mL水预洗,1 mL甲醇定容液过柱,开始接收洗脱液,5 mL甲醇洗脱,合并洗脱液,氮气吹干,1 mL甲醇复溶;冷冻净化操作为-20 ℃冷冻2 h以上,10 000 r/min离心5 min;过0.22 µm有机滤膜,上机检测。最终结果以Peak View软件结合本实验所建立数据库所筛查出的兽药化合物个数确定净化方法的优劣,结果见图2。

表2 174 种兽药化合物综合信息Table 2 Figures of merit of the method for 174 veterinary drug residues

续表2
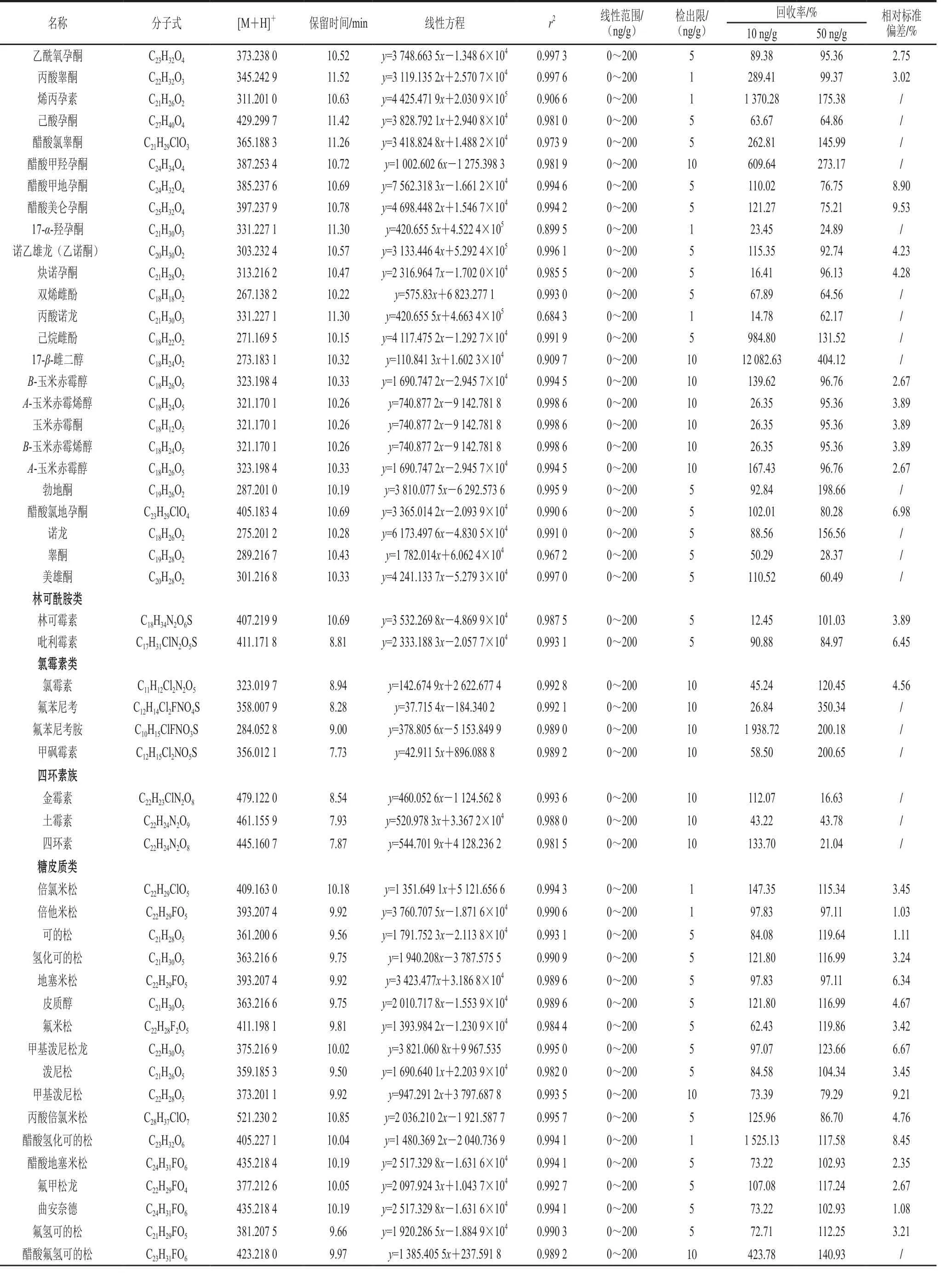
续表2

续表2
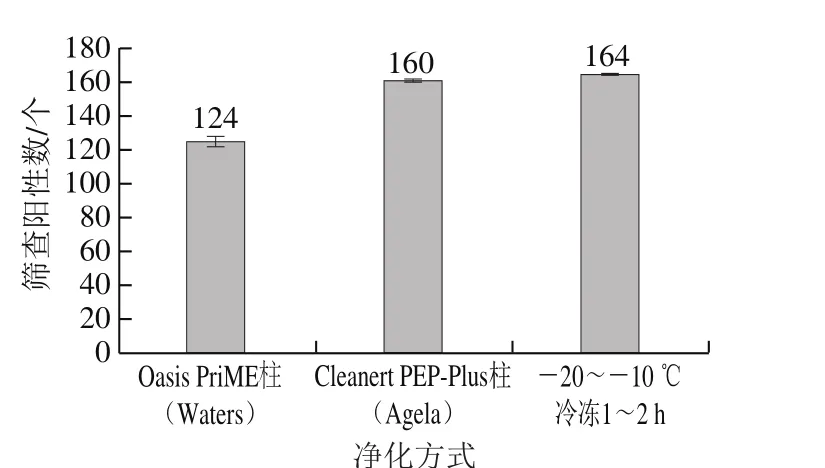
图2 净化处理方式对筛查结果的影响Fig. 2 Effect of different purification methods on the number of veterinary drugs screened
由图2可知,过柱净化后所筛查出的目标兽药化合物数目远低于冷冻净化后所筛查出的目标兽药化合物数目;此外,过柱净化[25,28-29]比冷冻净化耗时长,不利于后续大批量样品数据的采集;故选用冷冻净化作为本实验的除脂除杂技术。
2.3 提取试剂的优选结果
使用乙腈(或甲酸-乙腈)与乙酸乙酯按照1.3.1节步骤对目标化合物进行提取,分别使用两种净化方法(采用2.2节中优选的净化柱)净化后,供HPLC-TOF-MS/MS分析检测。通过对比总离子流色谱图中各化合物数量、以及可获得较好回收率(70%~130%)的目标分析化合物数量结果,结合Peak View软件筛查出的目标化合物数量结果,最终选出较优提取试剂,结果见图3。
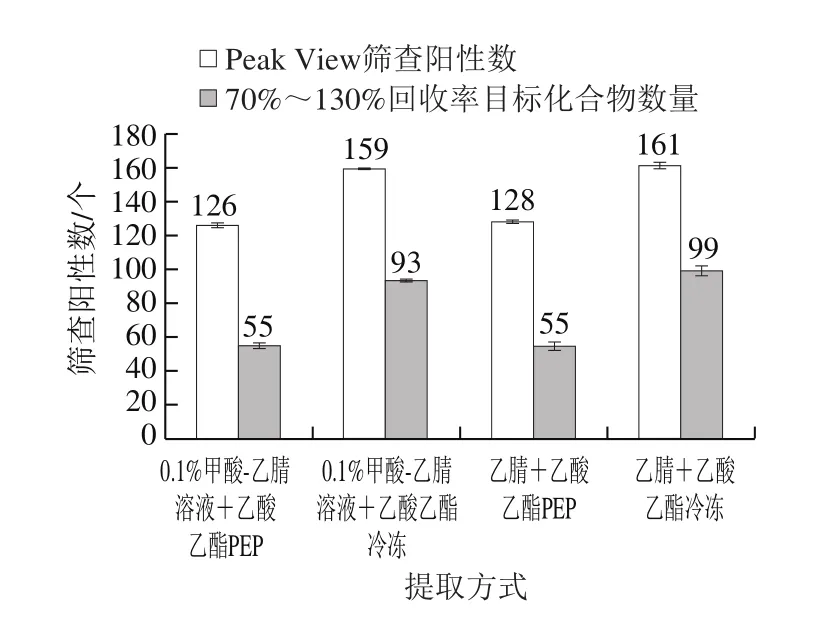
图3 提取处理方式对筛查结果的影响Fig. 3 Effect of different extraction methods on the number of veterinary drugs screened
由图3可知,无论是过柱净化还是冷冻净化,甲酸的加入没有明显提高对于目标化合物的提取,反而略有降低。未提取出的化合物主要是螺旋霉素、吉他霉素、氯霉素、金霉素和四环素等极不稳定的兽药。故最终选择纯乙腈与乙酸乙酯作为提取溶剂。
2.4 仪器条件的选择结果
2.4.1 液相色谱柱的选择
本实验结合实际应用,用200 ng/mL质量浓度的混合标准品溶液选择优化了3 种常用化合物分离色谱柱:ACQUITY UPLC®BEH HILIC柱(BEH柱,2.1 mm×100 mm,1.7 µm);ACQUITY UPLC®CSHTMC18柱(CSH柱,2.1 mm×100 mm,1.7 µm);Poroshell 120 EC-C18柱(P120柱,3.0 mm×150 mm,2.7 µm)。结果显示,从目标化合物峰的分离度看,CSH柱的效果优于BEH柱和P120柱,但是由于CSH柱和BEH柱中大部分出峰时间比较靠前,受溶剂影响比较大;而P120柱中目标化合物的出峰位置相对偏中,受溶剂影响相对较小,同时也方便后续液相洗脱程序的优化;此外,相对CSH柱和BEH柱,P120柱中色谱的整体响应强度更高,有利于充分的对目标化合物进行分析。故色谱柱最终选择为Poroshell 120 EC-C18柱。
2.4.2 流动相的选择
在前人总结的基础上,多种兽药筛查所用流动相主要是甲醇或乙腈与水。分别对甲醇和乙腈按照上述洗脱程序进行梯度洗脱,实验结果表明,两种有机溶剂的使用对多种兽药目标化合物的分离影响不显著,最后选择甲醇和水作为本实验的流动相。此外,经参考文献[30],发现甲酸的加入对于正离子扫描模式的峰形、峰的分离度,尤其是峰的响应强度有显著地改善,本实验也对其效果进行验证,见图4。
由图4可明显发现,加入0.1%的甲酸对于部分化合物的响应强度的确具有明显的提高,故本实验最终选择的流动相为0.1%甲酸溶液和甲醇。
2.5 生鲜乳中兽药残留筛查方法的验证结果
2.5.1 标准曲线与检测限
准确吸取上述200 ng/mL混合标准工作溶液0、125、250、500、750、1 000 µL于15 mL玻璃试管中,50 ℃氮气吹干,1 mL空白生鲜乳基质提取液复溶,过0.22 µm有机滤膜,上机检测。检测结果经Multi Quant软件分析,以质量浓度x作为横坐标,峰面积y作为纵坐标进行线性拟合,得线性方程。结果表明,绝大部分化合物在0~200 ng/mL范围内,各化合物面积与质量浓度之间呈良好的线性关系,其中,R2大于0.990 0的高达114 种。线性方程、线性范围、相关系数见表2。
取空白生鲜乳样品2 g,分别加入不同体积混合标准工作溶液制成含目标化合物1.0、5.0、10.0、20.0 ng/g的样品,涡旋混匀,避光静置30 min使其接触充分,与样品同法处理后进样测定信噪比。以RSN≥3时的浓度作为检测限,检测限在1~10 ng/g之间,符合检测要求,其检测限结果见表2。
2.5.2 回收率与精密度
称取同一来源的空白生鲜乳样品若干份,每份2 g,分别添加不同体积的混合标准工作液,制成10 ng/g和50 ng/g水平的质控样,按照样品同法处理后进样测定,结果带入标准曲线计算回收率与精密度。结果表明,在质控水平,本方法对绝大多数目标兽药化合物加标回收率较好:其中50 ng/g添加水平下,回收率在70%~130%之间的兽药化合物高达99 种;重复实验6 次,其相对标准偏差为1.03%~9.65%,见表2。
2.6 实际样品分析
本研究将建立好的HPLC-TOF-MS/MS方法用于实际生鲜乳中兽药残留分析,筛查来自上海不同牧场的50 个生鲜乳样,共有13 个样品兽药检出,涵盖糖皮质、磺胺、β-内酰胺、β-兴奋剂、类固醇类,结果见表3。

表3 实际生鲜乳兽药残留筛查结果Table 3 Screening results for veterinary drug residues in real milk samples
3 结 论
本研究利用乙腈和乙酸乙酯将目标兽药化合物从生鲜乳中提取出来,结合冷冻净化技术除脂除杂;采用优化的HPLC-TOF-MS/MS进行检测分析,建立生鲜乳中17 大类,174 种兽药的筛查方法,并将其应用到实际生鲜乳样品的兽药残留筛查。结果表明:方法简单快速,经验证具有较高的精准度和灵敏度,适用于复杂基质生鲜乳中高通量兽药化合物的筛查。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
煤化工(2022年3期)2022-07-08
色谱(2021年7期)2021-06-07
少儿科技(2021年6期)2021-01-02
河南畜牧兽医(2016年24期)2016-11-29
IT经理世界(2016年20期)2016-11-23
公民与法治(2016年4期)2016-05-17
中国资源综合利用(2016年10期)2016-01-22
IT经理世界(2014年8期)2014-05-05
云南畜牧兽医(2014年4期)2014-02-28
