
基于机器学习开发的危害性预测软件对罕见错义突变的预测评估
2018-11-19 06:46孙宇辉蒋廷亚连超群
皖西学院学报 2018年5期
党 孝,孙宇辉,蒋廷亚,周 阳,连超群
(1.美国费城儿童医院,宾夕法尼亚州 费城 19146;2.苏州奥根诊断,江苏 苏州 215000;3.江苏大学 生命科学研究院,江苏 镇江 212013;4.蚌埠医学院 医学临床检验诊断中心,安徽 蚌埠 230036)
新一代高通量测序技术的出现及其快速发展促进了遗传病致病基因的发现与临床分子诊断。在基因检测中,对患者基因组测序产生的大量候选突变位点中发现致病突变是科研以及临床分子诊断需要解决的关键科学问题。准确区分中性突变与有害突变对遗传病的临床检测有着重要的意义,目前的研究方法主要是参考突变的功能影响,群体突变频率信息,基因功能信息以及家系或多样本的位点验证信息[1]。研究表明,对于单个样本的外显子测序数据,即使过滤了群体常见等位基因频率(minor allele frequency(MAF)>1%)与位点功能,最终仍然有大约400个左右的罕见非同义候选突变位点[2,3]。
目前国内外对于突变的危害性预测已经开发出了多个不同的软件,从预测原理及预测方法上区分,其主要基于:1)蛋白质功能的改变:主要是突变引起蛋白质功能发生变化,如PolyPhen-2、SIFT、MutationTaster、FATHMM和 MutationAssessor等;2)进化保守性:主要是对多个物种核酸序列或蛋白序列进行多序列比对,分析同源序列的多态性,如GERP++、SiPhy、fitCons和PhyloP等;3)整合型软件:主要是利用机器学习等算法整合多个其他单独的预测软件为一整体分析预测,如CADD、fathmm-MKL、MetaLR、MetaSVM、VEST3、Eigen、DANN、GenoCanyon、REVEL和M-CAP等。
有研究表明整合型软件的预测效果整体优于单个软件[4],因此此类软件已广泛应用于位点的危害性预测。对于这些整合型突变危害性预测软件,由于其建立在不同的理念与算法基础上,基于不同的训练集,因此预测结果的准确性与特异性一直是需要评估的重点;评估结果也能更好地指导研究人员选择软件进行位点危害性预测。本研究收集两份独立的测试数据集和一份真实样本测序数据集对整合型预测软件(CADD v1.3[5],fathmm-MKL[6],MetaLR,MetaSVM[4],VEST3 v3.0[7],Eigen v1.1[8],DANN[9],GenoCanyon v1.0.3[10],REVEL[11]和M-CAP v1.0[12])进行评估,由于REVEL和M-CAP主要针对罕见错义突变的预测,为了公平评估,本文主要评测不同危害性预测软件对罕见错义突变的预测;在收集的致病突变位点中也发现错义突变占了致病SNP位点的绝大部分。
1 材料与方法
1.1 致病位点和中性位点测试数据集
收集两份独立测试数据集用于评测整合型突变危害性预测软件的效果,测试集1:ClinVar数据集,致病和中性的突变位点来源于ClinVar数据库[13,14](variant_summary.txt.gz文件,下载于ClinVar FTP,数据更新时间:09/10/2018),选取基因组为GRCh37的种系(germline)SNP位点,其中致病的SNP选取已报道临床意义为“Pathogenic”或“Likely pathogenic”的位点,同时为了保证致病位点的可靠性,过滤掉审查状态为没有明确证据显示是否致病的位点(no assertion criteria provided);中性的SNP选取报道临床意义为“Benign”或“Likely benign”的位点。测试集2:UniProt数据集,致病和中性的突变位点来源于UniProt/Swiss-Prot[15](数据更新时间10/10/2018),其中致病的SNP选取报道标签为“Disease”的位点,中性的SNP选取标签为“Polymorphism”的位点。由于UniProt数据库上记录的位点是氨基酸突变形式,因此使用TransVar[16]将氨基酸突变形式转化为GRCh37基因组坐标;如果测试数据集2中有出现数据集1的位点,则直接从数据集2中过滤掉此位点以保证两个数据集的独立性。
1.2 整合型预测软件对罕见错义突变预测评估
对于上述两份测试数据集,过滤只留下在公共群体数据库(1000 Genomes Project[2],Exome Sequencing Project(ESP)[17],Exome Aggregation Consortium(ExAC)[3]和UK10K[18])中次等位基因频率(MAF)<1%的罕见错义突变,然后分别使用CADD、fathmm-MKL、MetaLR、MetaSVM、VEST3、Eigen、DANN、GenoCanyon、REVEL和M-CAP对这些位点进行预测,预测效能的评估利用受试者工作特征曲线ROC(Receiver Operating Characteristic)以及ROC曲线下面积AUC进行比较,为了保证评估的公平性,选取了在所有要评估的预测软件上都有预测分值的突变位点进行ROC评估。ROC曲线的生成以及不同预测软件AUC值的计算使用R语言程序包“ROCR”[19]。
1.3 真实测序数据评估
除了上述两份独立的测试数据集,又收集一份真实的测序数据以评估这些软件在临床分子诊断上的效果。在EGA(https://www.ebi.ac.uk/ega/home)上申请来自于NIHR BioResource Rare Disease Consortium对遗传性眼病样本的测序数据(EGA号:EGAD00001002656,数据的bam文件来自于全外显子测序,CRAM文件来自于全基因组测序),这些样本中404位病人通过测序以及临床分子诊断已经找到其致病位点[20]。对申请下载的BAM或CRAM数据使用GATK v3.6 HaplotypeCaller call变异[21](只选择外显子区域Agilent SureSelect Human All Exon V5的变异位点),对call出的变异过滤“LowQual”以及深度小于5的SNP位点,然后对剩余的候选SNP位点使用M-CAP、fathmm-MKL、CADD、VEST3、REVEL、MetaLR和MetaSVM分别进行危害性预测。在这404份明确致病突变的样本中,筛选出致病错义突变位点,为保证评估的公平性,挑选所有致病的错义突变在上述要评估的预测软件中都有预测分值,最终得到118个致病的罕见错义突变,来源于123个眼科样本。
2 结果
2.1 致病位点的频率以及功能分布
致病突变的收集来源于ClinVar和UniProt数据库,最终在ClinVar中收集了30821个以及在UniProt中收集了14322个致病的SNP,致病突变在群体中的发生率低,因此大多为罕见突变。对这些致病突变在千人基因组1000 Genomes Project,Exome Sequencing Project(ESP)以及Exome Aggregation Consortium(ExAC)中次等位基因频率的分布也印证了这些致病位点中约99%以上都为罕见突变(MAF<1%)(图1);同时统计这些致病突变的突变类型,突变类型的注释基于Variant Effect Predictor(VEP)[22],发现错义突变占所有致病SNP的46%以上(表1)。错义突变占了单核苷酸突变类型的大部分,因此难以实现对疾病的致病性判断以及变异解读,根据美国医学遗传学与基因组学学会(ACMG)制定的序列变异解读指南,大多数错义突变不能明确分为致病或良性,只能判定为意义未明变异(VUS)[23],这对遗传病的临床诊断造成一定的困难,危害性预测软件的开发与实现可以辅助对位点致病性的判断,从而帮助研究人员发现疾病致病位点或者明确临床分子诊断。

图1 ClinVar和UniProt数据库中致病SNP频率分布图
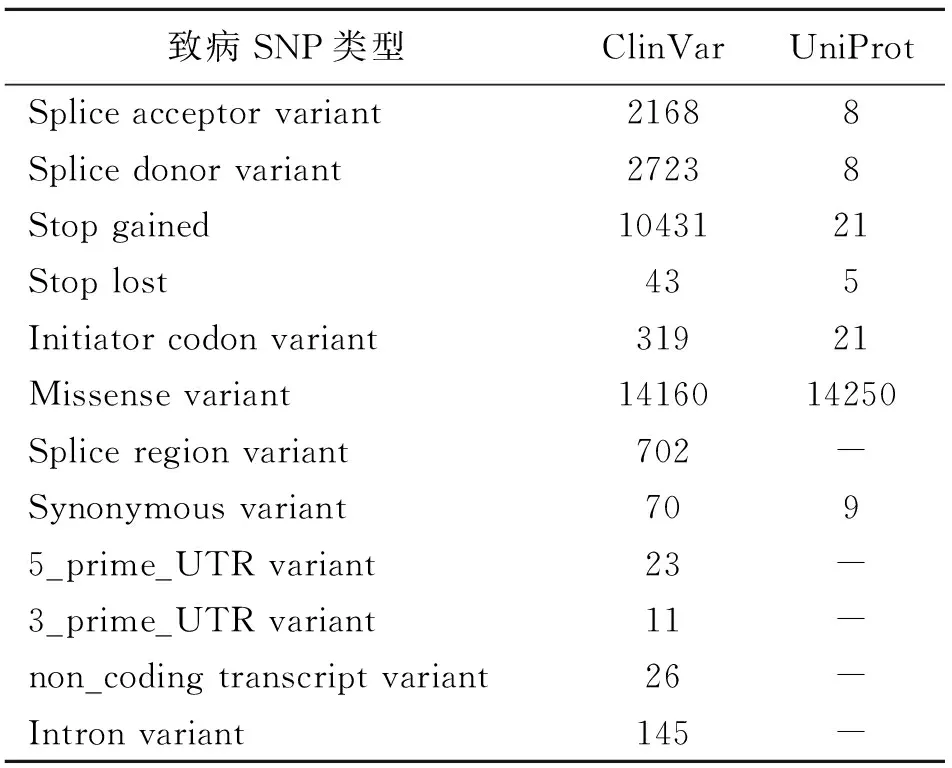
致病SNP类型ClinVarUniProtSplice acceptor variant21688Splice donor variant27238Stop gained1043121Stop lost435Initiator codon variant31921Missense variant1416014250Splice region variant702-Synonymous variant7095_prime_UTR variant23-3_prime_UTR variant11-non_coding transcript variant26-Intron variant145-

图2 测试数据集ROC曲线图((a)为ClinVar测试数据集结果;(b)为UniProt测试数据集结果,两个测试数据集相互独立)
2.2 整合型预测软件对ClinVar和UniProt数据库罕见错义突变预测
对收集的ClinVar和UniProt测试数据集,过滤群体频率与突变类型留下罕见错义突变位点,并确保所有位点在要评估的预测软件上都有预测分值。最终测试数据集ClinVar剩余12316个罕见错义致病突变和7988个罕见中性错义突变;测试数据集UniProt剩余11818个罕见错义致病突变和10427个罕见中性错义突变(附件1)。将上述两个测试集分别进行ROC曲线分析,通过ROC曲线以及曲线下面积AUC值的比较,综合两个测试集结果(图2(a) ClinVar测试数据集结果;图2(b) UniProt测试数据集结果),发现在罕见错义致病突变与中性突变的识别上,REVEL效果最好,准确性以及特异性明显优于其他软件,其他效果较好有M-CAP、MetaSVM、MetaLR和VEST3,其准确性与特性性高于剩余的5个软件。两个测试数据集是独立的,因此评估效果更能代表无偏性,避免评估软件在特定数据集上的过优或过差表现。
2.3 来源于123个眼科样本的118个罕见错义致病突变不同软件的预测
通过比较M-CAP、fathmm-MKL、CADD、VEST3、REVEL、MetaLR和MetaSVM在真实123个眼科测序样本的基因组罕见SNP危害性预测效果,同时重点关注这些样本的118个致病突变的预测结果(附件2)。统计这些软件对123个样本总共预测的罕见唯一的致病位点数目,同时这118个致病位点不同软件的预测结果分为两类:“Damaging”和“Tolerated”,比较发现尽管CADD和FATHMM-MKL对118个致病位点预测的Damaging数量多,但也将更多的中性突变预测为有害,因此假阳性率相对较高。REVEL、MetaLR和MetaSVM预测的Damaging数目少,但同时对其他中性突变预测为有害的数量低,因此在选择危害性预测软件时需要根据自己的研究需求,权衡假阳性率与假阴性率(图3,表2)。一个典型的单基因病患者一般携带1~2个致病突变,其余的大部分突变为中性突变[12]。

图3 不同软件对118个罕见错义致病突变的预测图(注:左边坐标表示致病突变数目,柱形图表示预测“Damaging”和“Tolerated”的数量;右坐标表示118个致病位点预测 Damaging的数量与123个样本中总共预测的致病的唯一突变数目的百分比值,由折线图展示)

软件有害阈值118个致病位点预测Damaging数目118个致病位点预测 Tolerated数目123个样本中总共预测的致病的唯一突变数目REVEL>0.589295,304MetaSVM>080385,420MetaLR>0.582366,160M-CAP>0.025115316,636CADD>201071125,394VEST3>0.5952313,695fathmm-MKL>0.5111728,462
3 讨论
在已发现的遗传病致病位点中很大一部分是罕见错义突变,区分突变的致病性对疾病致病基因的发现与分子诊断起着重要作用,本研究通过对基于机器学习开发的不同危害性预测软件使用两个独立的测试数据集进行预测评估,结果表明,REVEL效果最好,准确性以及特异性优于其他软件,其他效果较好的软件有M-CAP、MetaSVM、MetaLR和VEST3,尽管CADD、fathmm-MKL和DANN在罕见错义突变的预测上效果不突出,但他们对于非编码以及其他类型突变的危害性预测起着重要的作用,REVEL和M-CAP是最新开发的专门针对罕见错义突变的软件,因此在使用不同软件时需结合具体需求选择针对性的软件,辅助位点危害性的判断。本文也评估了部分软件在已明确致病突变的真实测序样本上的表现,结果提示在使用危害性预测软件时需要权衡假阳性率与假阴性率,而且预测结果只能作为对位点致病性的辅助判断,不能作为筛选或确定信息予以使用。
危害性预测软件的效果评估一直以来受到特定数据集以及突变类型的影响,研究表明不同的预测软件预测结果不一致率高,在不同的测试数据集上准确性与特异性波动大。不同的预测软件建立在不同的理念与算法基础上,基于不同的训练集,即使针对同一个位点,不同的软件对突变的危害性预测结果很不一致[24]。而且相同的软件对于不同的位点,不同的测试数据集预测结果的准确性与特异性波动大[25]。多个预测软件不同的预测结果会影响对突变危害性的判断,为了提升危害性预测软件的准确性,目前以更为具体的疾病,基因或通路信息研究是危害性预测软件提升的一个方向,如基于部分有突变热点或重要功能的基因的危害性预测软件开发[26];对于特定基因家族的危害性预测软件开发[27];基于不同的生物通路信息开发的危害性预测软件等。
4 致谢
感谢NIHRBioResource, University of Cambridge和NIHR BioResource Rare Diseases BRIDGE consortium允许下载使用他们测序的遗传性眼病样本数据。感谢费城儿童医院Center for Data Driven Discovery of Biomedicine提供访问交流机会。
猜你喜欢
广西医科大学学报(2022年5期)2022-06-07
昆明医科大学学报(2022年3期)2022-04-19
猪业科学(2021年3期)2021-05-21
矿产勘查(2020年3期)2020-12-28
生物学通报(2020年10期)2020-08-13
中南医学科学杂志(2019年6期)2019-12-05
科学与财富(2018年30期)2018-12-28
计算机应用(2016年9期)2016-11-01
新闻传播(2016年22期)2016-07-12
体育科技(2016年2期)2016-02-28
