
香茅草精油微乳液的构建及其抗氧化活性分析
2018-11-06 12:54陶紫赵振刚
现代食品科技 2018年10期
陶紫,赵振刚
(华南理工大学食品科学与工程学院,广东广州 510640)
香茅草(Cymbopogoncitratus (DC.) Stapf)属禾本科香茅属(Cymbopogon),是著名草本香种,含天然柠檬香味,主要分布在东半球热带及亚热带国家。我国的香茅属植物有8种,主要分布在南部地区[1]。因其具有和胃通气、健胃消脂和降压利尿等功效,香茅草在医药、日化、香精香料和饲料添加剂等行业应用广泛[2]。
香茅草精油 (Cymbopogoncitratus (DC.) Stapf essential oil,C-EO)具有广泛的药理活性:如,因具有一些香叶醛和橙花醛等醛类物质,常作为抑菌材料[3];有研究表明其主要成分中的柠檬醛物质具有较理想的抗氧化能力,其清除O2·,OH·有显著效果[4]。但因植物精油具有挥发性,对光线、氧气、温度敏感,且难溶于水,其在水环境中的运用受限。因此,本研究拟通过选用合适的表面活性剂、助表面活性剂和水相,在一定比例条件下制备精油微乳体系,使其具有较精油本身更稳定,更显著的抗氧化活性。
微乳液(Microemulsion),由水、油、表面活性剂和助表面活性剂在适当比例下自发形成,表现为透明或半透明、低粘度且各向同性[5]。因其稳定性好、光散射弱、可设计成高粘性或者凝胶状的流变性质,并且可用来包埋亲脂性活性成分的特点,微乳液在食品、制药和化妆品行业均有广泛的应用价值[6,7]。而香茅草因其生长周期短,管理粗放,投资小见效快,且其精油在国际市场上价格较高,因此是一种极具商业价值的常青草本作物。本文结合C-EO和微乳技术,制备香茅草精油微乳体系(Cymbopogoncitratus (DC.)Stapf essential oil Microemulsion,M-EO)来克服精油本身因稳定性差,难溶于水的弊端。通过绘制伪三元相图考察各因素对微乳液形成的影响来优化M-EO形成的工艺条件,寻找尽可能用更少的表面活性剂和助表面活性剂制备更稳定的微乳液。最后采用 DPPH,ORAC和PSC法测定并比较C-EO及M-EO的抗氧化活性,以期找到一种提高C-EO稳定性和增加其应用范围的方法,为拓展其商业化发展提供技术支持。
1 实验与方法
1.1 材料与试剂
植物材料:香茅草购于广州市一德路大型批发市场。
试剂:香茅草精油,自制;香茅草精油微乳液,自制;去离子水,自制;无水乙醇,广东光华股份有限公司;丙二醇,天津大茂化学试剂公司;甘油,天津启轮化学科技有限公司;Tween 80,Aladdin公司;Tween 60,Aladdin公司;Tween 20,Aladdin公司;2,2-二苯基-1-三硝基苯肼(DPPH),西格玛奥德里奇上海贸易公司;Trolox,Sigma公司;没食子酸,Sigma公司;2,7-二氯二氢荧光素二乙酯(DCFH-DA),Sigma公司;2,2’-偶氮二异丁基脒二盐酸(ABAP),Sigma公司;抗环血酸(Vc),Aladdin公司;荧光素钠,Sigma公司;氢氧化钾,国药集团;磷酸氢二钾,上海生工生物工程有限公司;磷酸二氢钾,上海生工生物工程有限公司;本实验中选用的其他试剂均为分析纯。
1.2 仪器与设备
水蒸汽蒸馏装置,常州普天仪器制造有限公司;气相色谱-质谱(GC-MS)联用仪(7890A-5975C DB-5ms),美国Agilent公司;气相色谱毛细管柱(30 m×0.25 mm×0.25 μm)(Basis),美国 Agilent公司;Hei-VAP Value旋转蒸发仪,德国 Heidolph公司;SHZ-D(Ⅲ)循环水式真空泵,巩义市予华仪器公司;BSA2202S电子天平,德国 Sartorius公司;MS-H280-PRO磁力搅拌器,大龙磁力搅拌器有限公司;DDS-11A 5424R电导率仪,上海雷磁创益仪器仪表有限公司;Nano-ZS纳米粒度仪(ZETASIZER-HT),英国Malvern公司;DU730分光光度计,美国Beckman Coulter公司;Filter Max F5多功能酶标仪,美国Molecular公司。
1.3 实验方法
1.3.1 C-EO的提取
参照魏娟[8]等提取姜黄精油的方法稍作修改,用水蒸气蒸馏法对香茅草进行蒸馏提取。采用水蒸汽蒸馏装置,将自然风干的香茅草剪切成5 cm左右长度,取10 kg原料于物料锅中,以1:10的料液比加入100 kg水蒸馏。收集冷凝液得精油粗提物后,用石油醚萃取,加适量氯化钠促进水油分层,取上层液,重复萃取 2次。将所有上层液用无水硫酸钠干燥,再用旋转蒸发仪除去石油醚得澄清透亮的金黄色香茅草精油,最后密封于棕色玻璃瓶中避光保存。
1.3.2 C-EO成分气相色谱-质谱联用(GC-MS)分析
采用安捷伦7890A-5975C气质联用仪;色谱柱:Agilent DB-5 ms(30 m×0.25 mm×0.25 μm);进样口温度:250 ℃;载气:高纯氦气(99.999%);柱流速:1 mL/min;进样量 0.2 μL;分流比 30:1;程序升温:50 ℃,保持 3 min,3 ℃/min升到 120 ℃,4 ℃/min升到180 ℃,15 ℃/min升到292 ℃,保持15 min。
质谱条件:电子轰击(EI)离子源,电离能量:70 eV;接口温度:280 ℃;离子源温度:230 ℃;四级杆温度:150 ℃;质量扫描范围:33~450 u;检索谱库:Nist08,最后用峰面积计算各成分占的百分比含量。
1.3.3 香茅草精油微乳液(M-EO)制备及优化
参照GUO Ruixue和MA Qiumin等[9,10]人制备微乳液的方法,并稍作修改。首先配置表面活性剂与助表面活性剂的混合液(混合活性剂,S+Cos),两者的质量比为Km,用磁力搅拌器搅拌均匀,静置平衡。再往混合活性剂中加入精油搅拌均匀作为油相,混合活性剂与精油的质量比表示为 Smix。随后往油相中边搅拌边滴加水来获得微乳液,通过绘制伪三元相图表明各配方组分形成微乳液的能力。(本实验意得到 O/W型微乳液,因此在优化微乳液制备工艺时只选择水相占比在50%及以上的样品作为实验对象。)
1.3.1.1 表面活性剂对M-EO形成的影响
分别用Tween 80,Tween 60,Tween 20作为表面活性剂,与无水乙醇配成 Km=4:1,3:1,2:1,1:1,1:0比例的混合活性剂,再配置 Smix=9:1,8:2,7:3,6:4,5:5,4:6,3:7,2:8,1:9 的油相。随后往油相中边搅拌边滴加去离子水,配成水分含量分别为50%,60%,70%,80%,90%的配方组分。静置平衡24 h,以4000 g的速度离心15 min,挑选出透明的样品,绘制伪三元相图。
1.3.3.2 助表面活性剂对M-EO形成的影响
以Tween 80为表面活性剂,分别以无水乙醇,1,2-丙二醇,甘油作为助表面活性剂,配成Km=3:1的混合活性剂。其他步骤同2.3.1。
1.3.3.3 Km值对M-EO形成的影响
以Tween 80为表面活性剂,以无水乙醇为助表面活性剂,配成 Km=4:1,3:1,2:1,1:1,1:0 的混合活性剂。其他步骤同2.3.1。
1.3.3.4 水相pH值对M-EO形成的影响
以Tween 80为表面活性剂,以无水乙醇为助表面活性剂,配成Km=3:1的混合活性剂。用0.1 M的盐酸溶液以及0.1 M氢氧化钠调节去离子水的pH分别为1.5,4.0,9.0,以之作为水相滴加到混合好的油相中。其他步骤同2.3.1。
1.3.3.5 水相中离子强度对M-EO形成的影响
以Tween 80为表面活性剂,无水乙醇为助表面活性剂,配成Km=3:1的混合活性剂。用氯化钠调节去离子水的离子强度,分别制成0.9%,2.0%的氯化钠溶液。用该去离子水滴加到油相中。其他步骤同2.3.1。
1.3.3.6 M-EO的构型分析和理化性质
电导率变化规律和粘度变化测定是判定微乳液构型的有效方法[11],本文测定Smix=8:2,Km=3:1时,微乳体系电导率和粘度随水分含量增加的变化规律。
将制备好的微乳液平衡24 h后,以4000 g的速度离心15 min。观察其是否分层或浑浊。用Nano-ZS激光粒度仪测定微乳液的粒径和分布(polydispersity index,PDI)。
1.3.4 C-EO及其微乳液抗氧化活性分析和比较
1.3.4.1 DPPH自由基清除能力的测定
参考 Wang等[12]人方法并稍微修改,测定 C-EO及M-EO对DPPH自由基的清除能力。用无水乙醇溶解DPPH粉末并稀释成2 mM的储备液。取2 mL不同浓度的C-EO和M-EO溶液加入2 mL经稀释后的DPPH储备液,涡旋均匀后,于室温下避光反应 30 min,在517 nm处测定吸光值Ai。以无水乙醇作为空白调零,以2 mL对应浓度的测试样的溶液加上2 mL无水乙醇作为对照(Aj)。2 mL无水乙醇加2 mL DPPH工作液的吸光值为Ac。则测试样对DPPH自由基的清除能力为:
DPPH自由基清除率(%)=[1-(Ai-Aj)/Ac]×100%1.3.4.2 氧自由基吸收能力(Oxygen Radical Absorption Capacity,ORAC)的测定
参照 Davalos[13]的方法,本实验用磷酸盐作为缓冲液(PBS,pH 7.4);以 Trolox为标准品。用 PBS配置系列浓度的 Trolox标准液(6.25,12.5,25,50 μM);样品C-EO和M-EO直接用PBS稀释。具体操作流程:在96孔黑色底部透明微孔板中依次加入20 μL抗氧化物质(Trolox或待测样品),于37 ℃环境下孵育10 min;随后往每个孔中加入200 μL荧光素钠溶液,孵育 20 min;随后每个孔中迅速加入 20 μL的ABAP溶液(空白对照孔加20 μL PBS);最后用多功能酶标仪测定各孔荧光度变化(激发光波长485 nm,发射光波长538 nm,每5 min测定一次,测定时长150 min)。所有测试样均做三次平行。
标品或者样品的荧光保护面积(Net AUC)等于标品或样品作用下的衰退曲线面积(AUC)减去空白对照作用的荧光衰退 AUC而得的荧光熄灭曲线的延迟部分面积(Net AUC=AUCsample-AUCblank)。样品的氧自由基清除能力ORAC值,由其荧光衰退曲线的保护面积与标品 Trolox的保护面积相比而得[ORAC=(Net AUCsample/Net AUCtrolox)×(Mtrolox/Msample)]。即,最后样品的ORAC值表示为每毫克样品的Trolox当量(μM Trolox equiv/mg,DW)。
1.3.4.3 过氧自由基清除能力(Rapid peroxy radical scavenging capacity,PSC)的测定
参照Adom等[14]对天然提取物过氧自由基清除能力的分析方法。以没食子酸和抗坏血酸为标准品,分别用磷酸盐缓冲液(PBS)稀释为5个系列浓度梯度;测试样为C-EO或M-EO,用PBS稀释成5个系列梯度;DCFH-DA在使用前用1 mM的氢氧化钾水解,并用PBS稀释。具体操作步骤为:在96孔黑色底部透明微孔板的微孔中依次加入 100 μL抗氧化物质(PBS(空白组),标准品或者测试样),100 μL水解后的DCFH-DA,50 μL ABAP溶液。最后用多功能酶标仪测定各孔荧光度变化(激发光波长485 nm,发射光波长538 nm,每5 min测定一次,时常40 min)。
样品的 PSC值为每组稀释液的荧光强度变化的时间动力曲线面积。以SA为标准品或测试样品的荧光积累量,CA为空白对照的荧光积累量,PSC值=1-(SA/CA)。以EC50值(即阻止50%荧光(PSC=0.5)产生所需要的测试样的浓度)作为最终比较抗氧化活性的指数。样品的抗氧化能力表示为μmol Vc equiv/g,DW。
1.3.5 数据统计分析
文中用origin 8.5软件绘制图表,Sigmaplot 12.5处理曲线面积积分,Calcusyn 2.0计算浓度效应,IBM SPSS21.0对数据进行 AANVOA显著性比较分析(p<0.05时,表明具有显著性差异)。所有实验均做3个平行,数据结果以平均值±标准差表示。
2 结果与讨论
2.1 C-EO的化学成分
采用水蒸气蒸馏法提取得到C-EO,利用GC-MS技术对精油进行成分分析,共检测出 M-EO具有 64种挥发性组分,占色谱馏分出峰总面积的96.60%。其主要成分如表1所示。其中相对浓度在1%以上的有16种,占总量的91.78%,其中超过60%成分为醇、醛或者烯类小分子物质。含量最高的4种成分别是(+)-香茅醛(35.69%),香叶醇(16.56%),D-芳香醇(12.47%),D-柠檬烯(3.77%),分别占其总量68.49%。
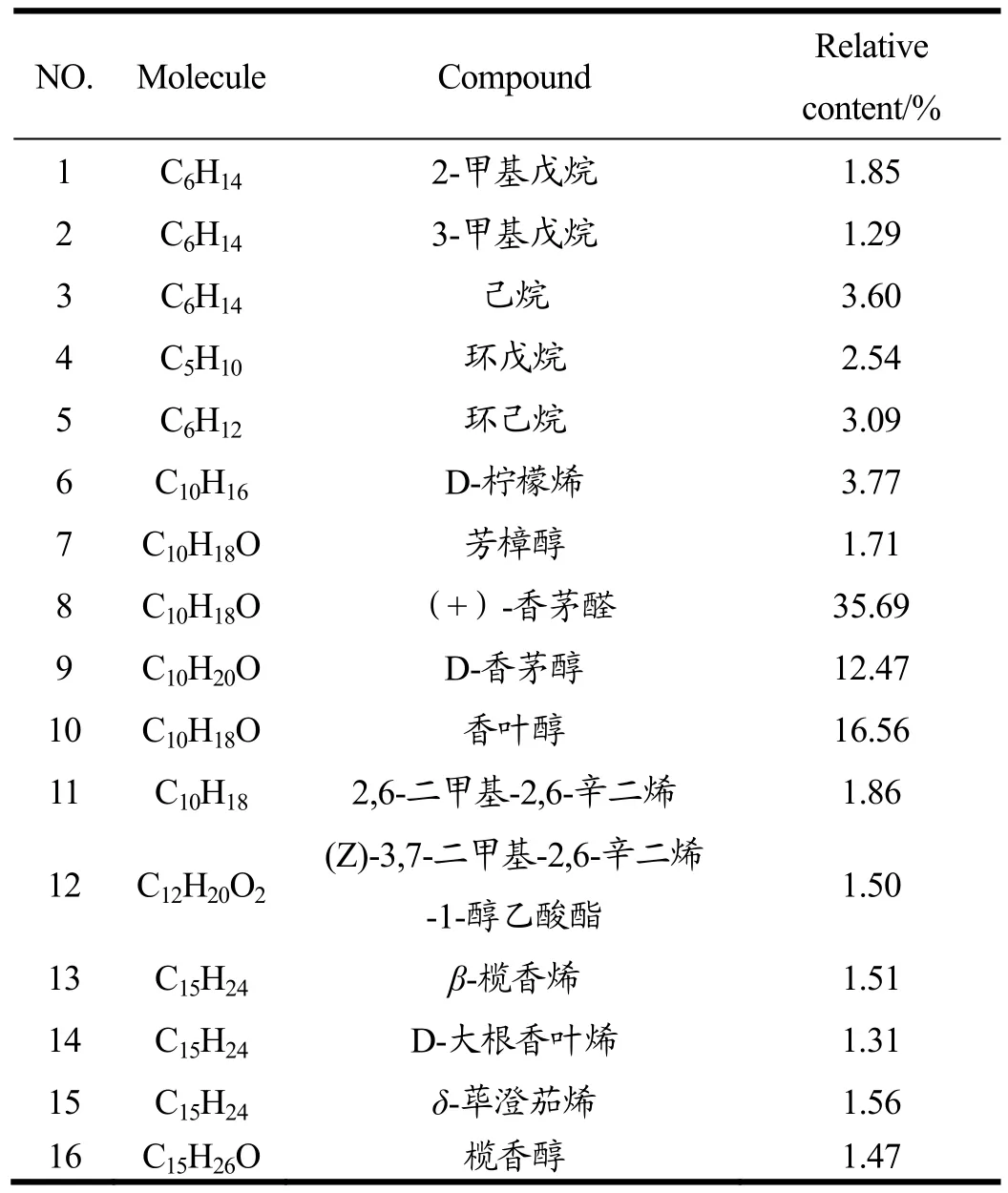
表1 C-EO挥发性成分中相对浓度在1%以上的化学成分Table 1 Chemical composition of C-EO in the relative concentration of more than 1%
由表1可知,本次研究对象的主要成分是一些醛、醇、烯和酯类物质。有报道表示,香茅草中含量较高醛醇物质,具有显著的抗菌活性,提高免疫细胞活性以及抗炎症作用[15,16]。更有一些动物实验研究表明,香茅草提取物可减少体内活性氧含量,提高血清总抗氧化能力,同时也可与其他成分作用产生协同效果提高总体抗氧化活性[17,18]。因此,香茅草精油成分分析为其之后的活性研究奠定了物质基础。
2.2 M-EO的制备和条件优化
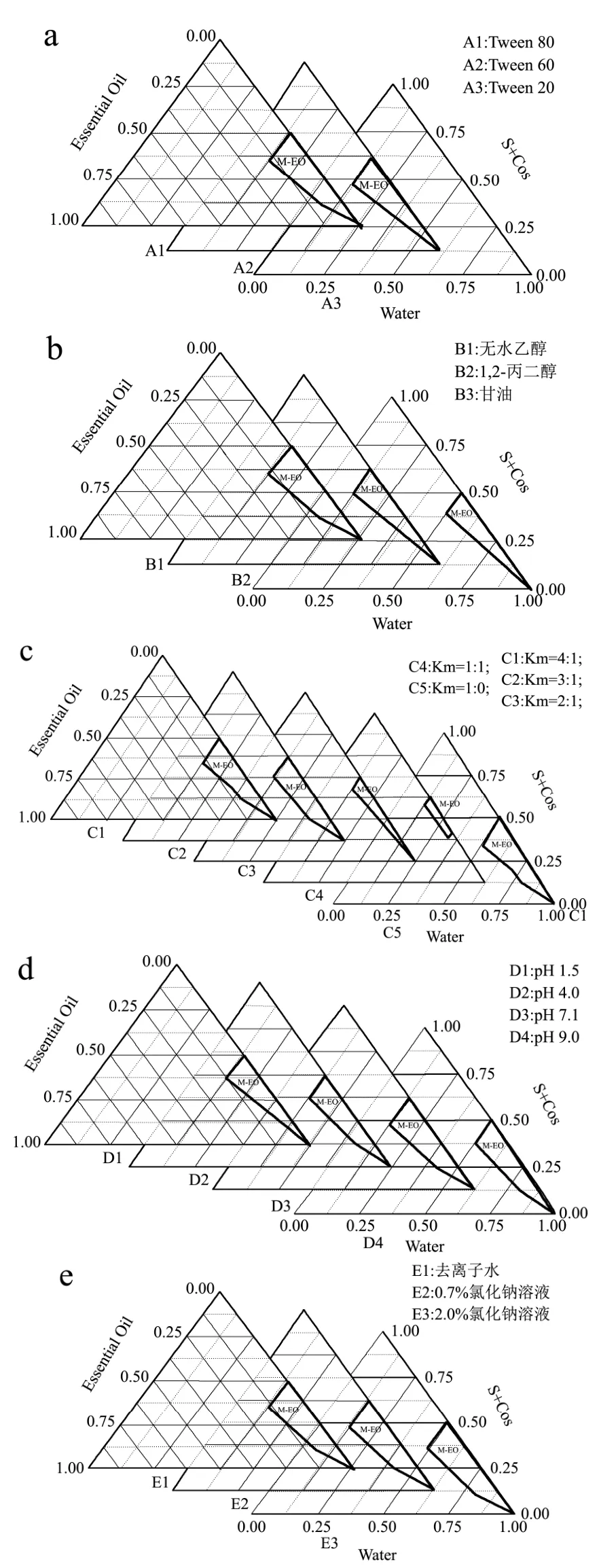
图1 各组成组分对M-EO形成的影响Fig.1 Effects of various components on pseudo-ternary phase diagrams
2.2.2.1 表面活性剂对M-EO形成的影响
表面活性剂是构成微乳液的基本物质,可降低界面张力促进微乳形成。因无毒、价格低廉和乳化效果显著等优势,Tween系列表面活性剂被广泛应用于食品、化妆品以及医药等工业领域[19]。本实验选用Tween 80,Tween 60,Tween 20作为研究对象,选出较优的表面活性剂。如图 1A 中显示,当 Km=3:1,Smix=8:2时,Tween 80的微乳区域面积大于Tween 60,而Tween 20在该体系中不能构成微乳液。该结果由各表面活性剂的理化性质决定:首先是 HLB值(Hydrophile Lipophilic Balance),HLB值反应表面活性剂中的亲水基团与亲油基团的平衡关系,值越高,亲水性越强。在已知选用的表面活性剂中,Tween 80的HLB值最大,Tween 60次之,Tween 20最小。因此,欲要获得O/W型微乳液,应选用Tween 80作为表面活性剂。
2.2.2.2 助表面活性剂对M-EO形成的影响
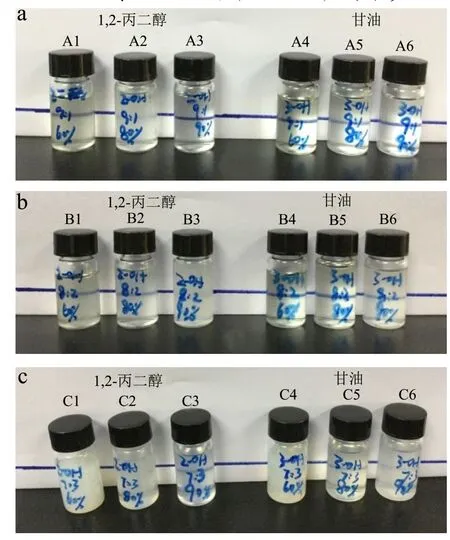
图2 以1,2-丙二醇和甘油为助表面活性剂的临界配方组分Fig.2 Sample figures in critical conditions with cosurfactants of 1, 2-propylene glycol and glycerinum
本研究选用无水乙醇,1,2-丙二醇,甘油作为助表面活性剂。由图1b的伪三元相图可知,无水乙醇对应的微乳区域面积最大,其余两者无显著区别。根据实际效果观察分析,甘油的助乳化效果稍强于 1,2-丙二醇(图2)。图2表示1,2-丙二醇和甘油制备成微乳液的一些临界条件的配方组成。其中 A2与 A5,B3与B6,C2与C5均是1,2-丙二醇和甘油在相同Km值,Smix值以及水分含量条件下的不同表现。从清晰透明度方面的对比可看出甘油的A5,B6,C5分别优于1,2-丙二醇同一条件下对应的A2,B3和C2。因此,实验结果表明无水乙醇优于甘油,1,2-丙二醇的效果最差。助表面活性剂的作用大小跟醇链本身的大小以及所带基团有关。Gradzielski[20]的报道表明,在非离子型O/W型微乳中加入短链醇可降低界面膜的弯曲系数,在水油界面上与表面活性剂形成混合吸附膜,降低表面张力,增加界面膜的柔性和流动性,减少微乳液形成所需的弯曲能;同时还可调节体系中的 HLB值以利于乳液形成。根据 Garti等[21]的解释,由于乙醇分子量和摩尔体积都较小,易于穿透微乳液第的栅栏层引起界面膜向油相凸起而利于微乳液的形成。而相同链长的短链醇,带有的羟基越多,其亲水性和极性越强,在一定条件下有利于亲水型微乳液的形成。
2.2.2.3 Km值对M-EO形成的影响
以Tween 80为表面活性剂,无水乙醇为助表面活性剂,按 Km=4:1,3:1,2:1,1:1,1:0 的比例混合,绘制伪三元相图,如图1C所示。由图可知,随着表面活性剂的占比升高,微乳区域面积增加。其中Km=4:1(图 1C1),1:0(图 1C5)的微乳区域面积最大,Km=1:1(图1C4)的微乳区域面积最小。这是由于Km值减小时,助表面活性剂含量进一步增加,油-水界面的张力降低,表面活性剂头基之间的吸引力减小,体系形成微乳液的能力减弱,从而微乳区域面积减小[22]。当 Km=4:1,3:1,1:0 时,三者的微乳区域面积相差不大,考虑产品最后应用的安全性和可适用性,优先选用表面活性剂更少的比例组分,因此本实验筛选出Km=3:1的比例。
2.2.2.4 水相pH值对M-EO形成的影响
以Tween 80为表面活性剂,无水乙醇为助表面活性剂,其中Km=3:1,Smix=8:2。用 0.1 M 的盐酸和 0.1 M的氢氧化钠溶液调节去离子水的pH作为水相。由图1D可知,直接用去离子水制备的微乳区域面积最大(图1D3),随着pH值越低,其区域面积越小。当pH值增加,体系呈现碱性,其微乳区域面积也受到影响而减小(图1D4)。该结果表明,中性的水相环境更利于微乳液的形成,王浩[23]在制备丁香油微乳体系时得到相似结果。
2.2.2.5 水相中离子强度对M-EO形成的影响
以Tween 80为表面活性剂,无水乙醇为助表面活性剂,其中Km=3:1,Smix=8:2。配置0.9%和2%的氯化钠溶液作为水相。其伪三元相图如图 1E所示,微乳区域面积随离子强度的变化相差不大,但是从临界配方组分的样品中观察可知(图3),实际上2%的氯化钠溶液样品的清晰透明度优于0.9%的样品,而去离子水的清晰度最弱。

图3 离子强度对M-EO形成的影响Fig.3 Effects of ionic strength on pseudoternary phase diagrams
图3中,在Km=3:1和Smix=8:2,水分含量为90%的条件下,A2与A3的透明度高于A1;同时,当Km=3:1和Smix=7:3,水分含量为90%时,B2和B3的清晰透明度明显优于B1。因此,在低离子强度下,随着离子强度的升高,体系的微乳化效果更好,这是由于低浓度的氯化钠溶液会迫使表面活性剂与水相分离,而与更多的油相形成反胶束,增加油相的载入,乳化效果更好。但是,当盐度过高,离子强度过大时,其对非离子型微乳液体系影响会较大。这是因为高浓度的盐溶液会产生盐析作用,降低表面活性剂的亲水能力,使其微乳形成能力减弱[24]。
2.3 M-EO构型的确定及其理化性质
2.3.1 M-EO构型的确定
图4表示当Km=3:1,Smix=8:2时,体系中电导率和粘度随水含量增加的变化趋势。
电导率的测定可以揭示微乳液体系的相变转化。由图4可知,体系的电导率在水含量小于30%时缓慢增加;当水含量在30%至60%时,电导率变化曲线斜率最大,随后增加速度减缓;当水含量大于70%,电导率有回落趋势。该变化规律表明,当水含量超过70%时,体系为O/W型微乳液。这是由于在低含水量的非离子型微乳液中,水相为内相,油相为连续相(W/O型)。电场作用下,小液滴在油相中泳动,碰撞时内核中的电解质离子穿过界面膜发生跃迁,随着电解质离子增加,离子跃迁次数增加,导致电导率逐渐上升。当水含量继续增加,液滴进一步膨胀,界面膜曲率增加,强度降低,电解质离子容易穿透,颗粒间相互距离接近,使得液滴间相互连接,形成导电链,电导急剧上升,使得电导率快速升高(B.C型)。接着导电链发展完善,随水含量继续增加,电导率的缓慢上升仅是由于增加电解质溶液的结果。当再增加水含量后,体系中导电离子是O/W型微颗粒,水分的增加导致体系不断被稀释,微乳液滴浓度降低,导致电导率下降[25]。
同样,图4中的粘度变化规律与电导率变化规律相似。当水分含量低于30%时,体系粘度缓慢增加;当含水量超过30%,粘度上升速度显著提升,在50%处达到最大值,随后急剧降低;当水分含量达到70%后,粘度变化变缓。一开始粘度的缓慢增加是由于W/O型液滴体积增加,液滴间相互作用力增大;随后的急剧增加则是因为随着水含量继续增加,分散颗粒增大,小球团形成,相互作用力增强(B.C型);突然的降低则是因为B.C型乳液向O/W型转变,此时以水相为连续相;水含量超过70%时,体系中的粘度变化则是由于更小的液滴的形成以及 O/W 型液滴的进一步稀释。通过电导率和粘度的测定可知,O/W型微乳液的水分含量在70%左右。
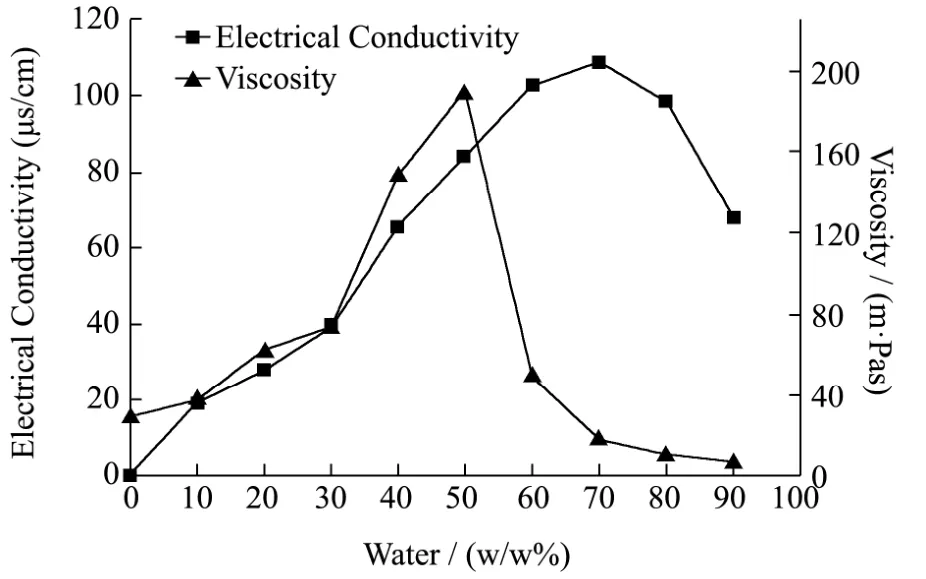
图4 M-EO体系电导率和粘度的变化曲线Fig.4 The electrical conductivity and viscosity curves of M-EO with different water content
2.3.2 M-EO的稳定性和理化性质
稳定性是微乳体系的重要参数,可作为该微乳体系质量的评价指标。制备好的微乳液在室温下平衡24 h,筛选出无可见浑浊或沉淀的样品。随后通过小型高速冷冻离心机在4000 g的条件下离心10 min,没有分层现象。
2.3.3 M-EO的理化性质
为了获得 O/W 型微乳液体系来改善精油水溶性差的问题以及进一步评估所制M-EO的性质,测定了Km=3:1,Smix=8:2,含水量为 70%配方组分样品的粘度,电导率,粒径,以及用去离子水稀释100倍和200倍后其粒径和PDI值的变化,其结果如表2所示。微乳液的原始粒径为16.50±0.53 nm,经过100倍、200倍稀释后,粒径均有小幅度减小。样品的PDI值变化规律显示原始样和稀释样的PDI值约为0.3,说明该微乳体系具有较理想的均匀性。由此判断,所选的Km=3:1,Smix=8:2,水分含量为70%的香茅草精油配方组分是较稳定、均匀、理想的微乳体系。
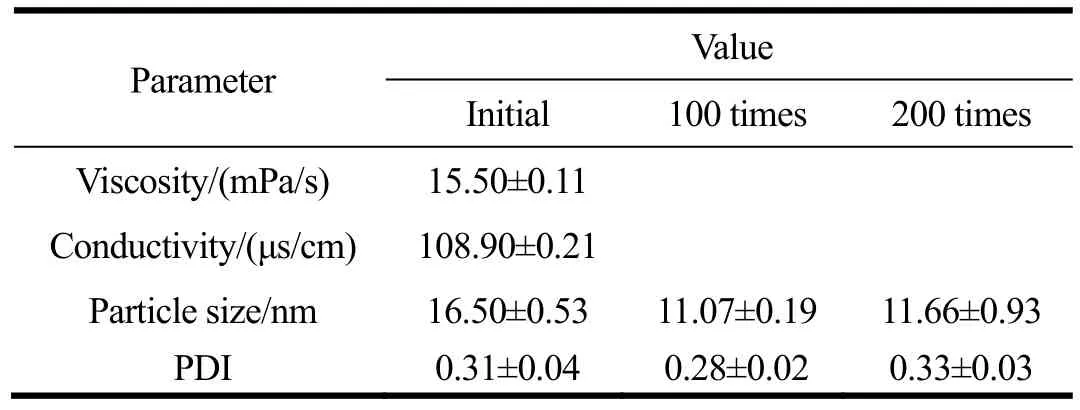
表2 香茅草精油微乳液的理化性质Table 2 Physicochemical parameters of M-EO
2.4 C-EO及其微乳液的体外抗氧化活性研究分析
2.4.1 DPPH自由基清除能力的测定
本实验选择 Km=3:1,Smix=8:2,水分含量分别为15%,50%,70%的微乳体系作为研究C-EO及其微乳液DPPH自由基清除能力的对象。
C-EO及其微乳液对DPPH自由基的清除能力如图5所示。图中表示C-EO及其不同水分含量M-EO的DPPH自由基清除率的EC50值,其中C-EO的EC50值约为0.26 mg/mL,显著低于其微乳液。表明C-EO的DPPH自由基抑制率优于其微乳液。在其三个配方组分的微乳液中,70%组分和 50%组分均显著优于15%的组分。同时,本实验也采用抗坏血酸作为阳性对照(图5B),以Vc当量表示DPPH自由基清除效果。其中C-EO的Vc当量约为162.88 μmol Vc equiv/g,70%和50%水分含量微乳的Vc当量相当,分别约为23.97 μmol Vc equiv/g,22.94 μmol Vc equiv/g,而 15%的组分活性最低。这可能是因为DPPH自由基清除实验以无水乙醇溶解样品和标准品,脂溶性的精油可直接发生反应,因此优于已组成颗粒的微乳液。而不同水分含量的微乳液的活性差异源于水分含量较高的组分,其颗粒较均匀,稳定性好;而70%配方组分与50%配方组分活性相当,主要是因为相对于O/W型的70%配方组分,脂溶性环境更利于50%配方组分(B.C型微乳液);15%水分含量的组分由于油相成分过高,其流动性和均匀度都低于水分含量较高的组分,因此DPPH自由基清除效果次于70%和50%的配方组分。
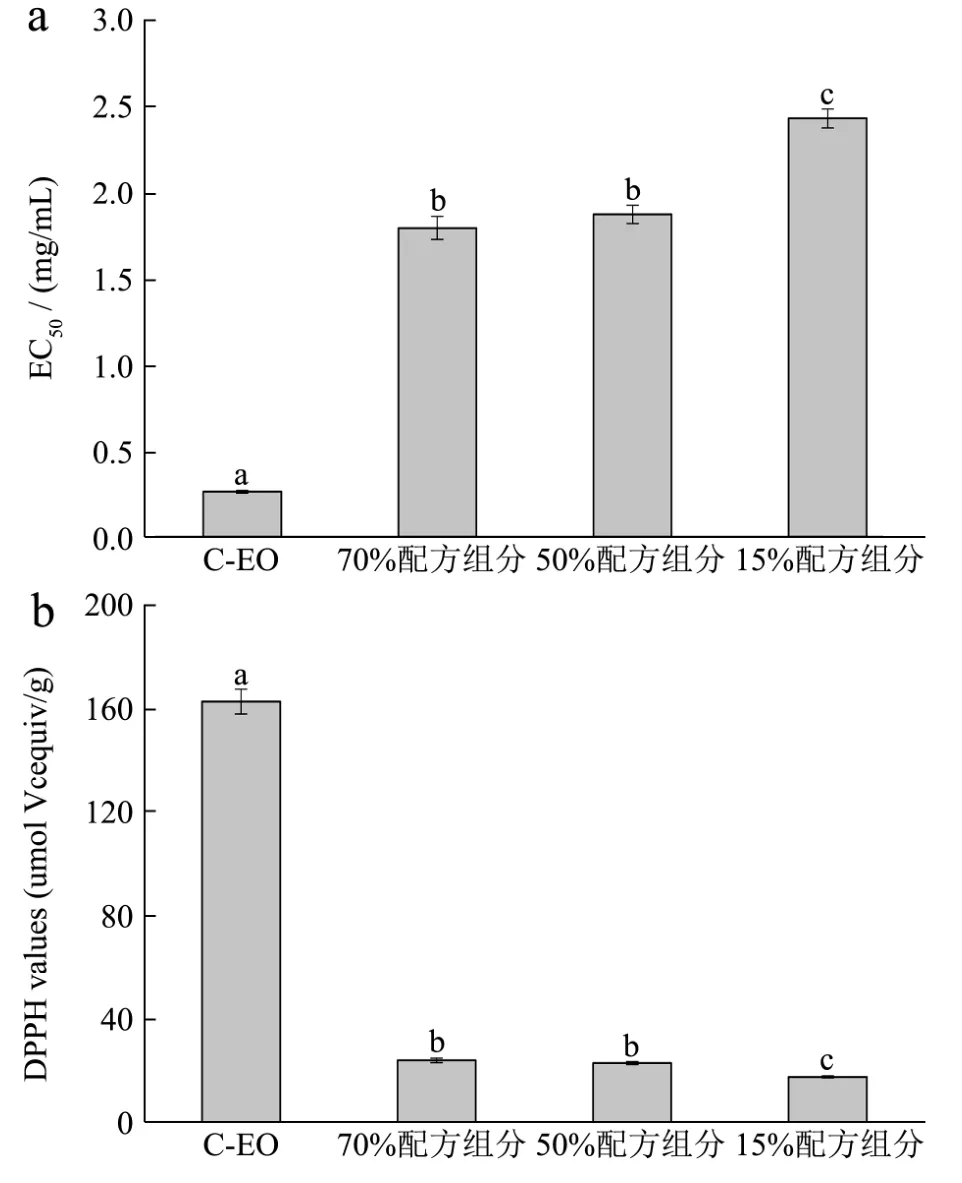
图5 C-EO及其微乳液的DPPH自由基清除能力Fig.5 The DPPH radical scavenging activity of C-EO and its microemulsions. (a) The EC50 value of DPPH radical scavenging rate. (b) The ascorbic acidequivalent of DPPH radical scavenging ability.
3.4.2 氧自由基和过氧自由基清除能力的测定(ORAC和PSC法)
由于本研究是为改善脂溶性精油在水溶性环境中运用,因此选择水溶性ORAC和PSC方法比较C-EO和M-EO的抗氧化活性,其结果如图6所示。
图6a表示C-EO及其微乳液的ORAC值,前者的ORAC值约为0.35 μmol Trolox equiv/mg,显著低于其微乳液的 ORAC值(70%配方组分:1.94 μmol Trolox equiv/mg;50%配方组分:1.11 Trolox equiv/mg)。类似的结果也在图6B中样品的PSC值中体现:C-EO的PSC值为1.53 Vc equiv/g,其微乳液中70%配方组分的PSC值达32.15 Vc equiv/g,50%配方组分达28.16 Vc equiv/g,即M-EO的活性均显著优于香茅草自身的抗氧化活性。香茅草精油微乳液对氧自由基和过氧自由基的清除能力高于其精油本身,主要是ORAC和PSC法是在水溶性体系中进行,而微乳液的形成克服了精油水溶性差的特点,提高了精油在水环境中清除自由基的能力。
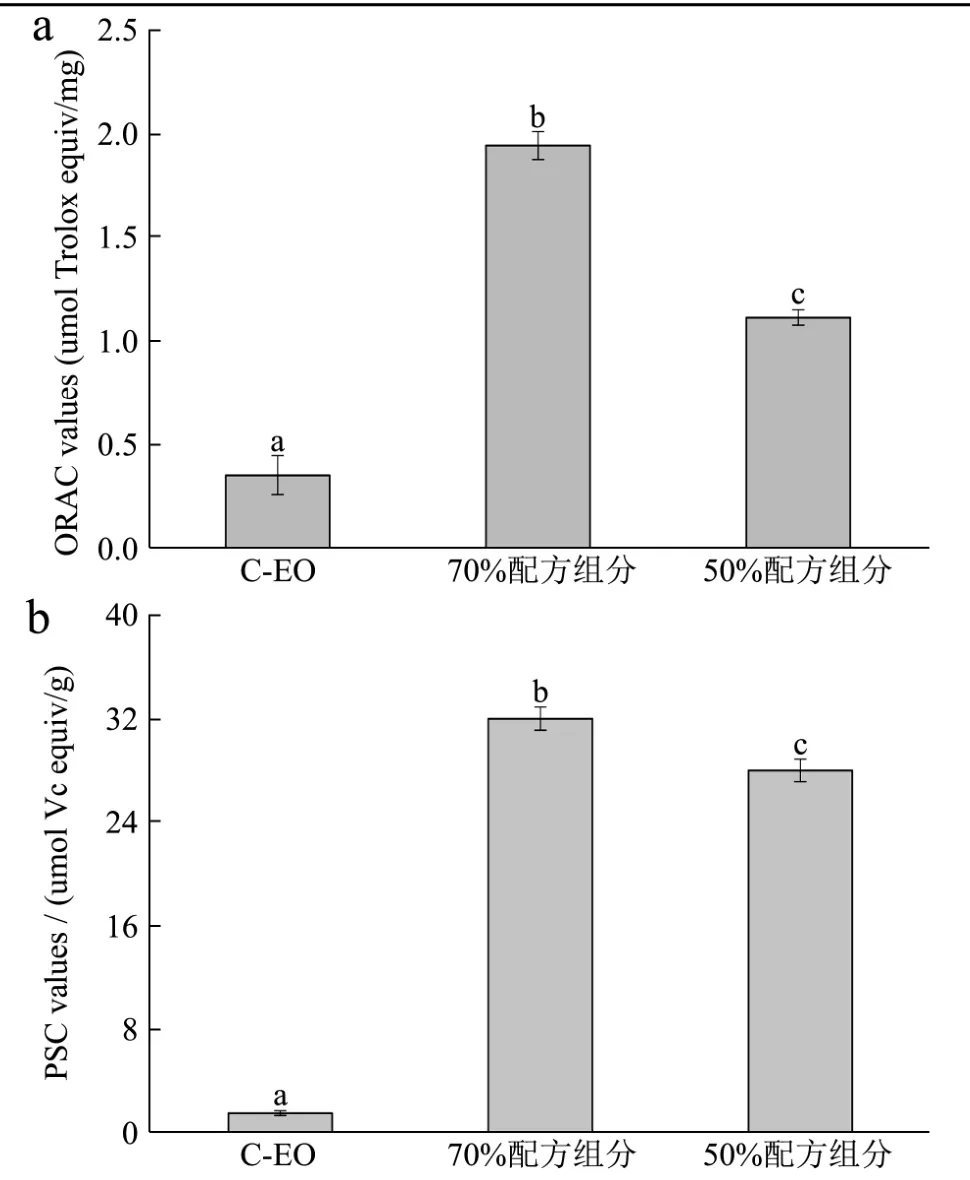
图6 C-EO及其微乳液对氧自由基和过氧自由的基清除能力Fig.6 The Oxygen radical and peroxyl radical scavenging capacity of C-EO and its microemulsions. (a) The ORAC value.(b) The PSC value
水分含量高的配方组分显示出更高的抗氧化活性是因为70%配方组分的M-EO是O/W型微乳液,相对于50%配方组分(B.C型)微乳液具有更高均一度的颗粒和均匀的体系,有利于样品发挥其活性作用。本文是首次采用ORAC值和PSC值方法评价香茅草及其微乳液的抗氧化能力的报道,因为ORAC和PSC方法均采用ABAP作为自由基,其产生的自由基性质更接机于生物体内产生的自由基,其可更好的分析抗氧化物质对由氧或过氧自由基引起的机体损伤的抑制,为今后开发香茅草精油为功能性食品或者药品提供理论基础[26]。
3 结论
本文采用水蒸馏法提取C-EO,并用GC-MS技术分析精油主要挥发性行成分。结果表明其主要物质有(+)-香茅醛(35.69%),香叶醇(16.56%),D-芳香醇(12.47%),D-柠檬烯(3.77%)。随后制备并优化精油的M-EO体系,最终挑选出由C-EO,Tween 80,无水乙醇,去离子水组成,且 Km=3:1,Smix=8:2,水分含量为70%的O/W型的稳定均一的理想微乳液。本研究最后通过 DPPH,ORAC及 PSC法分析比较C-EO及M-EO的体外抗氧化作用。结果显示,M-EO在水相环境中的抗氧化活性显著优于 C-EO,具有开发为天然抗氧化剂的潜力。本文构建的微乳液体系,解决了C-EO在应用过程中水溶性差等问题,拓展了其在食品、保健品和化妆品等领域的应用。关于精油其他生物活性是否增强,则需再进一步的研究。
猜你喜欢
发明与创新(2022年31期)2022-11-03
中国医药科学(2022年5期)2022-05-05
石油沥青(2019年5期)2019-11-16
中成药(2017年7期)2017-11-22
优雅(2016年2期)2016-06-03
中国果菜(2016年9期)2016-03-01
中国继续医学教育(2015年6期)2016-01-07
中国洗涤用品工业(2015年11期)2015-02-28
分析化学(2014年6期)2014-07-10
天然产物研究与开发(2014年8期)2014-04-27
