
UPLC-MS/MS法测定面粉中草甘膦残留量的不确定度评定
2018-11-01 08:15王紫昕冯秀娟
现代食品 2018年17期
◎ 王紫昕,朱 捷,冯秀娟
(宁夏食品检测研究院,宁夏 银川 750000)
草甘膦(Glyphosate)纯品为非挥发性白色固体,大约在230 ℃熔化,并伴随分解。25 ℃时在水中的溶解度为1.2%,不溶于一般有机溶剂,其异丙胺盐完全溶解于水。不可燃、不爆炸,常温贮存稳定,是一种非选择性、无残留灭生性除草剂,对多年生根杂草非常有效,广泛用于种植行业。其残留含量的检测原理是通过试样用水提取,经固相萃取柱净化,与9-芴基甲基三氯甲烷衍生化反应后,用液相色谱-质谱/质谱测定,内标法定量。
1 实验参数与条件
1.1 设备、试剂
材料:各大商超及小型店铺售卖的面粉。设备:超高相液相色谱-质谱联用仪(Waterstqsmicro)。标准品:草甘膦标准品99.0%;氨甲基膦酸标准品98.0%;同位素内标1,2-C13N15标准溶液,100 μg/mL。
试剂:二氯甲烷(AR)、甲醇(AR)、盐酸(AR)、9-芴基甲基三氯甲烷(FMOC-CL)≥99.0%、丙酮(AR)。
1.2 检测方法
1.2.1 液相色谱洗脱条件

表1 液相色谱洗脱条件表
1.2.2 质谱条件
离子源,ESI;模式,正模式;检测方式,MRM;干燥器温度:325 ℃;干燥器流量:7 L/min;喷雾器压力:275.8 kPa;EMV(+),300;鞘气温度:350 ℃;鞘气流量:11 L/min。
1.2.3 标准使用液
准确称取各标准物质于10 mL聚乙烯容量瓶中,用水溶解,加入2滴盐酸,并用水定容至刻度,配制为1.0 mg/mL的标准储备液。
准确吸取浓度为100.0 μg/mL的1,2-13C15N草甘膦内标标准溶液1.0 mL至10 mL聚乙烯容量瓶中,用水定容至刻度,配制成10.0 μg/mL的同位素标准工作液。
准确吸取适量混合标准储备液和同位素内标工作溶液,用水配制成5.0、10.0、20.0、50.0、100.0、200.0 ng/mL的混合标准工作液,其中同位素内标浓度为25 ng/mL。
1.3 样品前处理
称取5 g样品于100 mL离心管中,加入0.25 mL内标工作液,加入40 mL水、20 mL二氯甲烷,高速匀质2 min,4 000 r/min离心5 min,取上清液于100 mL聚乙烯容量瓶中,残渣再加入40 mL水重复提取一次,合并上层水溶液并定容至100 mL,带净化。C18小柱用5 mL甲醇、10 mL水活化后加入上述水溶液,弃去1~2 mL初滤液后接收。准确移取1.0 mL滤液与标准溶液一同加入200 μL 5%硼酸盐缓冲液,混匀,加入200 μL 1.0 g/L的FMOC-Cl溶液,混匀,40 ℃下衍生化反应,放置过夜,衍生后的溶液过滤膜,供超高相液相色谱-串联质谱仪测定。
2 不确定度的来源
2.1 数学模型的建立
面粉中草甘膦含量测定按公式(1)计算:

式(1)中,X为面粉中草甘膦的含量,mg/kg;C为由标准曲线得出的样液中待测物的浓度,ng/mL;C0为试剂空白,ng/mL;V为试样定容体积,mL;m为试样的质量,g。
2.2 标准溶液浓度引入的不确定度
草甘膦标准不确定度:

2.3 标准储备液配制过程中称量引入的不确定度
草甘膦标准储备液。

氨甲基膦酸标准储备液。
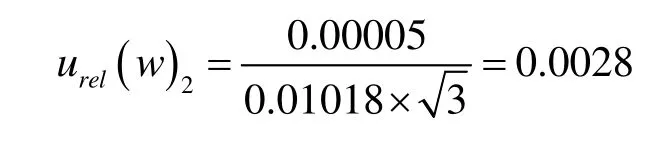
2.4 稀释过程引入的不确定度
标准工作曲线是由准确吸取适量混合标准储备液和同位素内标工作溶液,用水配制成5.0、10.0、20.0、50.0、100.0、200.0 ng/mL的混合标准工作液,其中同位素内标浓度为25 ng/mL。
绘制标准工作曲线过程中使用了一系列玻璃量具,按照JJG 196-2006《常用玻璃量具检定规程》的要求,均有相应的最大允差,按照三角分布考虑,k=,由此估算相对不确定度分量,见表1。

表1 标准系列配制过程中量具校准引起的不确定度表
草甘膦标准系列配制中引入的不确定度:

2.5 线性拟合得出质量浓度的不确定度
草甘膦最小二乘法拟合标准溶液质量浓度-峰面积结果,见表2。

表2 草甘膦最小二乘法拟合标准溶液质量浓度-峰面积结果表
采取面粉提取液测试10加标回收值,方程求得的平均质量浓度为D1=21.18 ng /mL时,则D1的标准不确定度:

式(2)中:

标准系列的平均浓度水平;

则X0的相对标准不确定度:

最小二乘法拟合氨甲基膦酸标准溶液质量浓度-峰面积结果,见表3。

表3 最小二乘法拟合氨甲基膦酸标准溶液质量浓度-峰面积结果表
本例中对面粉测定液进行了10次加标回收测定,由直线方程求得的平均质量浓度为D2=27.16 ng/mL时,则X2的标准不确定度:

式(3)中:

标准溶液的平均浓度;
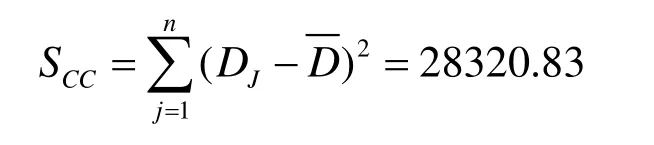
n为18;N为10次。
则X2的相对标准不确定度:
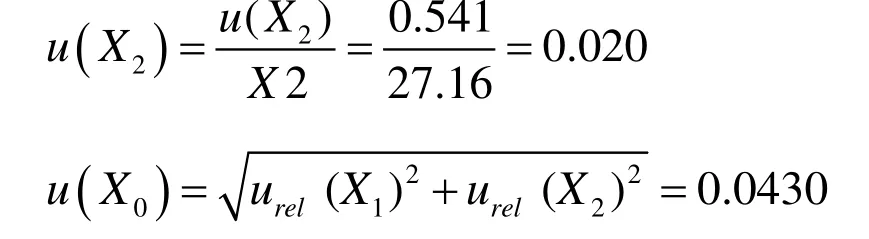
2.6 样品制备
2.6.1 取样
按SN/T 1923-2007的规定,准确称量至0.01 g,称样量为5.0 g,JJG 1036-2008《电子天平检定规程》规定,两次称量天平不确定度:
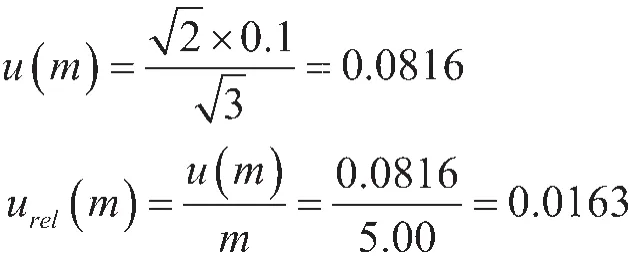
2.6.2 定容体积的标准不确定度
称取5 g样品于100 mL离心管中,加入0.25 mL内标工作液,加入40 mL水、20 mL二氯甲烷,高速匀质2 min,4 000 r/min离心5 min,取上清液于100 mL聚乙烯容量瓶中,残渣再加入40 mL水重复提取一次,合并上层水溶液并定容至100 mL,带净化。C18小柱用5 mL甲醇、10 mL水活化后加入上述水溶液,弃去1~2 mL初滤液后接收。准确移取1.0 mL滤液与标准溶液一同加入200 μL 5%硼酸盐缓冲液,混匀,加入200 μL 1.0 g/L的FMOC-Cl溶液,混匀,40 ℃下衍生化反应,放置过夜,衍生后的溶液过滤膜,供超高相液相色谱-串联质谱仪测定。
100 mL容量瓶校准引入的标准不确定度为:

1 mL刻度试管由校准引入的标准不确定度为:
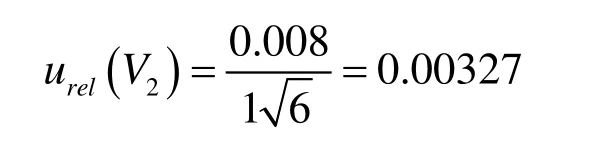
1 000 μL移液器由校准引入的标准不确定度为:

1 000 μL移液器由校准引入的标准不确定度为:
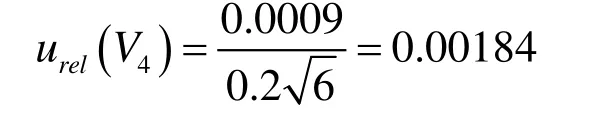
2.6.3 样品制备过程中的不确定度

2.7 复现性
同等条件下,对草甘膦试样中所含含量进行了10次独立性检测,结果见表4。
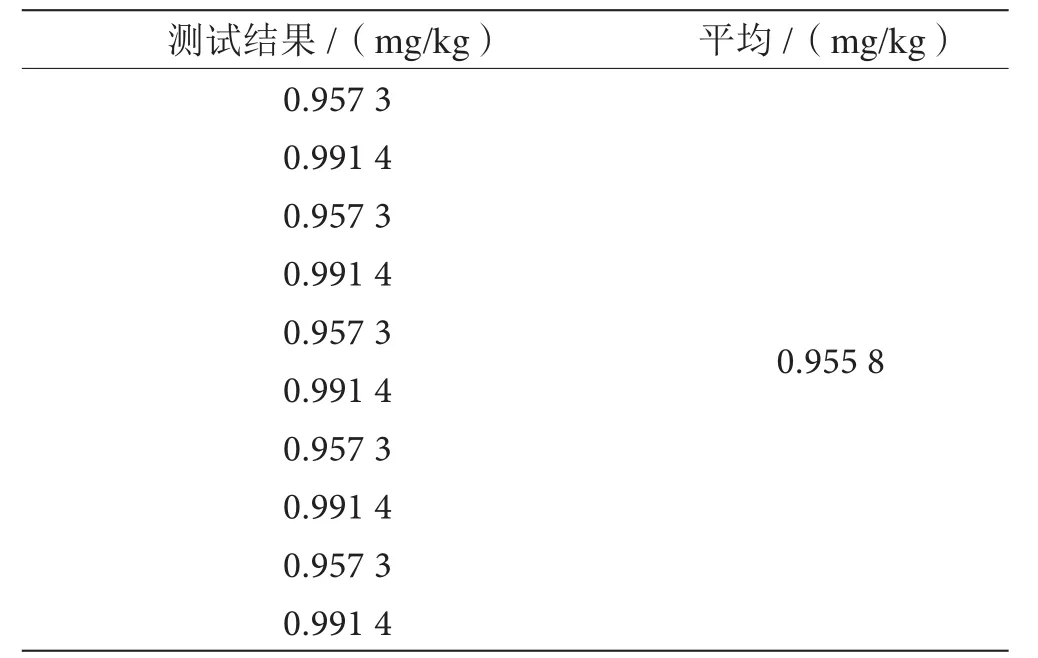
表4 草甘膦含量10次测定结果表
在同等条件下,对草甘膦样品进行10次检测,其含量算数平均值为:

单次测定的标准偏差为:
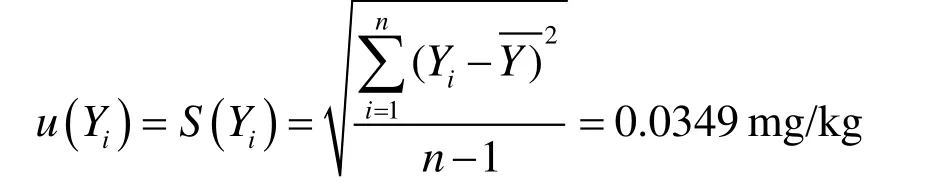
算数平均值不确定度:

3 草甘膦各不确定度来源结果
草甘膦含量不确定度来源结果表,见表5。

表5 草甘膦含量不确定度来源结果表
3.1 合成标准不确定度
面粉中草甘膦测量的合成标准不确定度基于公式计算如下:

3.2 扩展不确定度
试样中草甘膦的扩展不确定度的取K=2,扩展不确定度为:
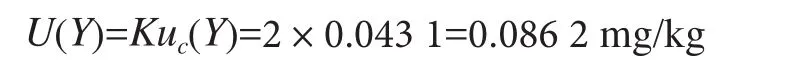
相对扩展不确定度:

结果:当K=2时,Y=(0.955 8±0.086 2)mg/kg
4 结论
本实验通过对面粉中草甘膦残留量不确定度的来源进行分析,并对各个分量进行了精细的计算,从结果可以发现,由于实验需采取衍生法进行实验,在衍生化的过程中,线性相关性的不确定来源相对较大,在同等条件下的复现性测试过程中所引起的不确定度次之。所以,通过草甘膦的不确定度的计算,可以促使在实验过程中应该更加精细化、精准化操作,使草甘膦残留量的不确定度降低。此外,在实验过程中,草甘膦的不确定来源分析与结果,提示工作人员在操作过程相对复杂的实验中,需操作人员的耐心与专业才能更好地完成实验。总的来说,本方法为更好地进行食品中草甘膦残留量的测定提供了有效的数据分析与结论。
猜你喜欢
中国氯碱(2022年10期)2022-11-22
化学工程师(2020年7期)2020-09-07
农药科学与管理(2019年6期)2019-11-23
山东工业技术(2019年6期)2019-03-27
世界农药(2019年2期)2019-01-06
制造技术与机床(2017年9期)2017-11-27
科技创新导报(2017年20期)2017-09-13
通信电源技术(2016年5期)2016-03-22
中国氯碱(2015年5期)2015-06-15
营销界(2015年23期)2015-02-28
