
PGRN和Rev-erbβ双基因敲除HEK 293细胞系的构建及应用
2018-10-25 09:11:36陈芳杨沛艳朱久玲
生物工程学报 2018年10期
陈芳,杨沛艳,朱久玲
1 安康学院 现代农业与生物科技学院,陕西 安康 725000
2 陕西师范大学 生命科学学院基因治疗研究室,陕西 西安 710062
对目的基因组特定位点进行打靶修饰技术,也称靶向基因组编辑技术,是基因工程研究的重点,也是研究基因功能的主要手段之一。目前,该技术被广泛有效地应用于构建细胞和动物疾病模型、培育动植物新品种以及治疗单基因遗传性疾病。靶向基因组编辑技术最早由Mario Capecchi于 20世纪 80年代提出的[1],是基于自然状态下DNA同源重组(Homologous recombination,HR)方式可以对基因进行定点编辑(敲除或敲入) 的作用,由于在自然状态下同源重组的效率低、大约只有百万分之一,因此很大程度上限制了此项技术的应用。自21世纪以来,科学家相继研发出了多种核酸酶来介导靶向基因组编辑技术的高效运行。其中较早应用的基因组编辑核酸酶有锌指核酸酶(Zinc finger nuclease,ZFN) 技术和转录激活因子样效应物核酸酶(Transcription activator-like effector nucleases,TALEN) 技术[2-5]。利用这两种核酸酶在基因组上对靶位点进行特异性切割导致双链 DNA的断裂,进而会引发细胞的同源重组或非同源末端连接(Non-homologous end joining,NHEJ)的方式修复受损 DNA,来实现基因定点的缺失(敲除)、插入(敲入) 以及基因修正等多种突变,极大地提高了靶向基因组编辑的效率,并使基因组编辑技术得到了迅速发展。2013年初,出现的规律成簇间隔短回文重复序列/Cas核酸酶(Clustered regularly interspaced short palindromic repeats/associated nuclease9,CRISPR/Cas9) 系统,是一种不依赖于FokⅠ核酸酶的新型基因组编辑技术[6-7]。该技术是由古菌和细菌基因组中串联重复序列(CRISPR/Cas) 获得性免疫系统发展而来的[8]。经科研人员改造后的 CRISPR/Cas9系统只需将 Cas9和单链引导 RNA(singleguide RNA,sgRNA)两种成分导入哺乳动物细胞中就可进行基因组的精确编辑,以实现靶基因定点的插入、缺失等突变修饰作用。CRISPR/Cas9技术具有构建容易、操作简单和周期短等明显的优势,作为一种新型有效的靶向基因组编辑技术,近年来有许多关于CRISPR-Cas9技术在多种细胞和生物个体中成功实现了基因组编辑操作[9-15]。目前,CRISPR/Cas9靶向基因组编辑技术不仅广泛应用于模式生物(如细菌、酵母、果蝇、斑马鱼、小鼠、大鼠、猪和猴等) 的基因编辑中,而且也应用于许多哺乳动物正常和肿瘤细胞系的基因研究中,具体研究内容涉及细胞或动物模型的构建、基因功能研究、基因转录调节及基因治疗研究等,已成为基因功能研究、寻找药物作用靶点的重要工具[12,16-22]。
颗粒蛋白前体(Progranulin,简称PGRN),又被称为蛋白内皮素前体(Granulin-epithelin precursor,GEP)、PC细胞源的生长因子(PC cell derived growth factor,PCDGF) 和内皮素前体(Proepithelin)等,是由593个氨基酸残基组成的多功能分泌性糖蛋白分子[23-25]。PGRN作为一种多功能细胞生长因子,广泛参与机体多种生理、病理调节功能,如生长发育、组织损伤修复、炎症反应、糖脂代谢以及肿瘤的发生等过程[26-28]。大量研究结果表明,PGRN不仅是一个多功能的免疫调节因子,在炎性反应中参与调节多种信号通路发挥促炎症或抗炎症的作用[29];PGRN还是一种重要的脂肪细胞因子,在脂肪组织中参与调节白细胞介素6(IL6) 进而介导高脂诱导小鼠产生胰岛素抵抗,并促进糖尿病、肥胖等疾病发生[30];另外,PGRN还是一个肿瘤调节因子,在许多肿瘤细胞中过度表达,参与肿瘤细胞的增殖、浸润以及血管生成等过程。但截至目前PGRN参与机体生理、病理调节功能的机制研究还不完善[27,31]。因此,为了深入研究PGRN生物学功能,前期利用酵母双杂交系统(Yeast two-hybrid systems) 筛选实验研究发现PGRN可能与核受体Rev-erbβ分子存在相互作用。Rev-erbβ是核受体Rev-erbs家族成员之一,它作为机体重要的转录因子可以直接结合在靶基因启动子 RORE(puG/AGGTCA)位点上,对靶基因转录进行调控。已有研究表明,脂肪代谢有关因子 Srebp-1c和载脂蛋白 ApoCIII等都是核受体 Rev-erbβ的靶基因[32-33]。为研究PGRN与 Rev-erbβ相互作用可能存在的生理意义,本实验利用CRISPR/Cas9基因打靶技术尝试构建PGRN和Rev-erbβ双基因敲除的HEK293细胞系,并在该细胞系中初步研究 PGRN和Rev-erbβ相互作用是否影响Rev-erbβ对靶基因转录的调控。总之,构建的PGRN和Rev-erbβ双基因敲除HEK293细胞系为进一步深入研究PGRN与Rev-erbβ相互作用的生理意义提供了必要的研究工具。
1 材料与方法
1.1 材料
1.1.1 质粒与细胞株
大肠杆菌Escherichia coli1-T1感受态细胞由陕西师范大学基因治疗研究室制备并保存;克隆载体pGEM-T Easy购自Promega公司;Cas9表达载体质粒购自Addgene公司。慢病毒pLenti/CMVLoxp-Linker-Loxp-EF1α-Puro 、 pLenti/CMV-Cre-Hygromycin的骨架质粒载体由本实验室构建保存。HEK 293人胚肾细胞株购自美国典型培养物保藏中心(American type culture collection,ATCC)。Rev-erbβ 基因敲除的 HEK293(Rev-erbβ-/-)C3-6细胞系由本实验室构建并保存。
1.1.2 试剂与仪器
质粒DNA小提中量提取试剂盒、血液/细胞/组织基因组提取试剂盒均购自天根生化试剂公司;DNA回收试剂盒(AXYPrep DNA GelExtraction Kit) 购自Corning公司;X-treme GENE HP DNA Transfection转染试剂购自Roche公司;Prime ScriptTMRT Reagent Kit(Perfect Real Time)反转录试剂盒、SYBR®Premix EXTaqTMⅡRT-PCR试剂盒以及实验用到的限制性核酸内切酶、T7E1酶、LATaqDNA聚合酶购自 TaKaRa公司;兔抗人PGRN单克隆抗体(mAb)购自Abcam公司;小鼠抗人源 GAPDH单克隆抗体购自Proteintech公司;实验中所用到的引物合成、核酸序列测序均由杭州金唯智公司完成;Master cycler PCR仪、微量移液器、5415D微型高速离心机购自Eppendorf公司;蛋白、核酸电泳系统、785型半干法转膜仪购自 Bio-Rad公司;超低温–80 ℃冰箱、CO2培养箱、1285型生物安全柜购自Thermo公司;E-25型细菌培养摇床购自美国NBS公司;生化培养箱购自上海精宏实验仪器公司;凝胶成像仪购自上海复日公司;全自动化学发光成像系统购自上海天能公司;高压灭菌锅购自日本SANYO公司;Thermo Scientific Varioskan Flash全波长多功能酶标仪购自美国 Thermo公司。
1.2 方法
1.2.1 相关引物设计与合成
依据 PGRN蛋白质的结构功能域,用(http://crispr.mit.edu) 网站在PGRN基因第5、6个外显子的区域中设计了4个不同的针对PGRN基因的 sgRNA,即 PGRN sgRNA1–4。并在每条PGRN sgRNA 序列中的正义链 5ʹ端添加上ACCG,反义链的 5ʹ端添加上 AAAC,使其分别能与经BsaⅠ酶切的pU6-sgRNA back bone表达载体(sgRNA表达载体) 形成互补的黏性末端。所设计的PGRN sgRNA寡核苷酸序列结果见表1。这4个PGRN sgRNA主要针对于PGRN基因G、F、B功能域。并根据PGRN的打靶序列片段(Target sequence fragment,TSF)设计出了4条打靶鉴定引物,PCR外巢检测引物 sp1/sp2和内巢检测引物sp3/sp4,具体引物序列见表1。

表1 PGRN sgRNA寡核苷酸及用于打靶检测PCR引物序列Table1 The oligonucleotide sequences of PGRN sgRNA and PCR amplification primers for knock-out PGRN detection
1.2.2 pU6-PGRN sgRNA1–4载体的构建及PGRN sgRNA活性筛选
1) pU6-PGRN sgRNA1–4载体构建
将金唯智公司合成的特异的 PGRN sgRNA1–4寡核苷酸序列,各取15 µL两两配对、混匀,室温放置1–2 h变性退火。取退火后的产物 4.5 µL与经BsaⅠ酶切的 pU6-sgRNA 骨架载体相连,获得pU6-PGRN sgRNA1–4表达载体。将获得的阳性质粒载体送金唯智公司测序、正确后,可用于后续PGRN sgRNA打靶效率的检测。
2) pU6-PGRN sgRNA1–4活性检测
将上述构建成功的4.0 µg的PGRN sgRNA表达载体和对照载体,分别与6.0 µg的Cas9表达载体用脂质体转染法共转染到HEK293细胞,72 h后收集转染细胞,提取细胞基因组。用提取的基因组为模板,用LATaq酶进行PCR扩增PGRN的TSF区域(PCR外巢扩增引物sp1/sp2,内巢扩增引物 sp3/sp4,PCR扩增程序和条件参考文献[34]),PCR扩增产物再经变性退火、凝胶回收获得PGRN 打靶区DNA片段。取500 ng上述回收的DNA片段,用0.5 µL的T7E1酶(约5 U活力单位),37 ℃酶切20 min,酶切产物用1.5%琼脂糖凝胶电泳分析sgRNA活性。
1.2.3 携带Cas9和PGRN双sgRNA的慢病毒打靶载体的构建
依据上述PGRN sgRNA活性检测的结果,参照文献[15]将2个有活性的sgRNA串联到一起可以显著提高打靶效率。用文献[15]的方法,选择2个具有较高打靶活性的单个PGRN sgRNA,构建出携带PGRN双sgRNA的pU6-PGRN-double sgRNA的表达载体。用SpeⅠ和NotⅠ酶切PGRN双sgRNA表达载体(pU6-PGRN-double sgRNA),获得PGRN-double sgRNA酶切片段,用ClaⅠ和SpeⅠ酶切 Cas9 表达载体质粒获得 Cas9基因片段。将上述片段先后与NheⅠ和NotⅠ酶切以及SfuⅠ和XbaⅠ酶切的慢病毒 pLenti/CMV-Loxp-Linker-Loxp-Puro骨架载体相连,获得含重组hCas9和PGRN double sgRNA的慢病毒载体(具体载体中的主要元件为 pLenti/CMV-Loxp-Cas9-sgRNA2-U6-sgRNA3-U6-Loxp-EF1α-Puro,图 1)。
1.2.4 细胞培养及转染
HEK293细胞、HEK293 C3-6细胞均用DMEM培养基进行培养,另外添加10%的胎牛血清、10 g/LL-谷氨酸、丙酮酸钠、100 μg/mL链霉素和100 IU/mL青霉素。37 ℃、5.0% CO2的培养箱中静置培养。将HEK293细胞或HEK293 C3-6细胞接种至24孔细胞培养板中培养,当细胞密度达到 70%-80%时,用 X-treme GENE HP DNA Transfection转染试剂转染目的质粒DNA到细胞中,转染具体的操作方法参照产品说明书。
1.2.5 PGRN稳定敲除的HEK293细胞系的建立
在文献[34]Rev-erbβ基因敲除的 HEK293(Rev-erbβ-/-)C3-6细胞系的基础上,利用携带Cas9和PGRN double sgRNA的慢病毒对PGRN基因进行打靶敲除。首先,将构建成功的含重组Cas9和PGRN double sgRNA的慢病毒载体(pLenti/CMV-Loxp-Cas9-sgRNA2-U6-sgRNA3-U6-Loxp-EF1α-Puro)(图 1),转染到 HEK293T 细胞中进行慢病毒包装。再取包装后 48 h的细胞培养液上清,感染 Rev-erbβ基因敲除的 HEK293(Rev-erbβ-/-)细胞。感染 48 h后,用 1.2 µg/mL 嘌呤霉素筛选,待筛选稳定后(7-14 d),收细胞基因组进行打靶检测。然后,用pLenti/CMV-Cre-Hyg慢病毒感染有打靶效果的细胞(以除去整合到打靶细胞基因组中Loxp-Cas9-sgRNA2-U6-sgRNA3-U6-Loxp)。再用(0.2 µg/mL潮霉素B) 筛选,待细胞筛选稳定后,收集打靶细胞的基因组。采用PCR扩增重组到基因组中的Cas9基因,检测打靶细胞中Cas9基因的清除情况,并对清除完全的打靶细胞进行克隆化操作。提取阳性单克隆细胞的基因组,用外、内巢PCR检测引物P1/P2和P3/P4,PCR扩增TSF区基因片段,将获得扩增目的片段与野生型TSF区序列相比较,并分析打靶细胞的PGRN基因突变类型。将PCR扩增的产物含有突变型的(PGRN TSF) 序列进行测序,由测序结果确定阳性克隆细胞是否是基因敲除(插入、缺失)的单克隆(PGRN-/-) 细胞株。

图1 PGRN慢病毒打靶载体结构示意图[35]Fig.1 Schematic illustration of the targeting vector for knock-out of human PGRN[35].
1.2.6 PGRN基因敲除的HEK293细胞系的鉴定
1) PCR对PGRN基因敲除细胞系的鉴定
取对照HEK293 C3-6细胞和PGRN基因敲除的HEK293细胞,用基因组提取试剂盒分别提取上述细胞的基因组。以提取的基因组为PCR扩增模板和特异的外、内巢引物P1/P2、P3/P4为打靶鉴定的PCR引物,采用巢式PCR对PGRN基因打靶区(TSF区) 基因片段进行扩增,PCR反应及扩增条件参见文献[34]略有改动。取少量 PCR扩增产物,用1.5%琼脂糖凝胶电泳初步检测分析打靶敲除后PGRN基因TSF区基因突变型类型。并将PGRN基因敲除的HEK293细胞不同的PCR扩增产物分别进行凝胶回收、纯化,连接pGEM-T载体,提取阳性克隆质粒送测序。再将PGRN TSF序列片段的测序结果与对照细胞PGRN TSF序列进行比对,来对PGRN基因敲除细胞系的PGRN基因的突变类型进行鉴定分析。进一步确定PGRN基因敲除单克隆阳性细胞系PGRN基因的突变类型。
2) qRT-PCR检测PGRN mRNA水平
取一定量对照和PGRN基因敲除的HEK293细胞,用 TRIzol处理分别提取细胞的总 RNA。取500 ng细胞总RNA作模板,用TaKaRa公司的反转录试剂盒反转录成 cDNA。再用 SYBR®Premix ExTaq试剂和特异的PGRN RT-PCR引物sp5/sp6扩增不同细胞的PGRN基因,以甘油醛-3-磷酸脱氢酶(GAPDH) 为内参,每个反应设3次重复。根据qRT-PCR检测结果,通过比较基因敲除细胞与对照细胞的PGRN mRNA表达水平的变化,确定PGRN基因敲除细胞系的PGRN mRNA表达水平的变化。
3) Western blotting检测PGRN蛋白质水平
取对照、PGRN基因敲除HEK293细胞60 mm平皿各一盘,用200 μL RIPA(含蛋白酶抑制剂)的细胞裂解液提取细胞总蛋白质。用二辛可宁酸(Bicinchonininc acid,BCA) 定量试剂盒测定细胞总蛋白质浓度。分别取50 µg总蛋白量的对照和PGRN敲除细胞蛋白质提取液上样,经 10%SDS-PAGE分离。再经转移电泳将电泳分离的蛋白条带转移到PVDF膜上,PVDF膜用5% BSA(W/V) 的PBST溶液室温封闭1.0–1.5 h;用兔抗人源 PGRN单克隆抗体(1∶1 000) 和小鼠抗人源 GAPDH 单抗(1∶500) 分别与印迹膜室温共孵育1-1.5 h;1×PBST洗涤孵育后的PVDF膜3次(5 min/次);然后用HRP标记的山羊抗兔IgG(1∶10 000稀释) 和山羊抗小鼠IgG(1∶10 000稀释)的二抗孵育液与 PVDF膜室温孵育 1 h;分别用1×PBST和1×PBS彻底洗涤印迹膜3次(5 min/次);最后用化学发光法检测目的蛋白质的印迹条带,确定PGRN基因敲除细胞系中PGRN蛋白质的表达水平。
1.2.7 PGRN介导 Rev-erbβ对靶基因启动子转录活性调控的检测
在 PGRN和 Rev-erbβ双基因敲除 HEK293 C3-6/C23细胞系中,通过回补PGRN和Rev-erbβ来研究 PGRN介导 Rev-erbβ对靶基因启动子ApoCIII和 Srebp-1c转录活性调控的具体作用。操作方法如下:将实验室前期构建的Rev-erbβ靶基因启动子荧光素酶报告基因载体pGL3-hApoCIII promoter-Luci和pGL3-hSrebp-1c promoter-Luci 150 ng分别与100 ng pAd5 E1-CMV-hPGRN质粒(人源PGRN真核表达载体)或/和100 ng pAd5 E1-CMV-hRev-erbβ质粒(人源 Rev-erbβ真核表达载体) 以及20 ng pRL-CMV-Renilla Luci质粒(内参载体) 共转染到24孔PGRN和Rev-erbβ双基因敲除细胞系中,用空载体pAd5-E1质粒补充使每组实验转染的质粒总量为370 ng,转染后48 h收集细胞总蛋白。另外以转染同样量的pAd5-E1空载体作为对照组。用双荧光素酶(Dual-luciferase)试剂和全波长多功能酶标仪检测各实验组的荧光素酶相对活性,反映出PGRN与Rev-erbβ相互作用介导Rev-erbβ对hApoCIII和hSrebp-1c启动子启动荧光素酶转录活性的影响情况。
2 结果与分析
2.1 PGRN sgRNA活性检测与筛选
将 pU6-PGRN sgRNA1、pU6-PGRN sgRNA2、pU6-PGRN sgRNA3和pU6-PGRN sgRNA4的表达载体和对照载体,分别同重组hCas9表达载体共转染到HEK293细胞中,72 h后收集转染细胞,提取其基因组。用 PCR扩增 PGRN打靶区 TSF片段,琼脂糖凝胶纯化、并回收目的DNA片段,再用T7E1酶切回收的DNA片段,经1.5%琼脂糖凝胶电泳分析得出PGRN 4个sgRNA的打靶活性(图 2)。与对照质粒相比较,pU6-PGRN sgRNA2和pU6-PGRN sgRNA3有明显的切开条带,条带大小与预期的结果相一致,表明pU6-PGRN sgRNA2和pU6- PGRN sgRNA3有较高的打靶活性。将有活性的PGRN sgRNA 2和sgRNA 3串联,构建携带双PGRN sgRNA和Cas9的pLenti/CMVLoxp-Cas9-sgRNA2-U6-sgRNA3-U6-Loxp-EF1α-Puro慢病毒载体,对 HEK293 C3-6细胞进行PGRN基因打靶操作。
2.2 PGRN基因敲除的HEK293细胞系的建立与鉴定
用包装好的携带Cas9和PGRN sgRNA2+ sgRNA3的慢病毒感染 HEK293(Rev-erbβ-/-) 细胞对 PGRN基因进行打靶敲除。病毒感染后48 h,向培养基中添加1.2 μg/mL嘌呤霉素进行筛选,筛选10 d细胞稳定后,收细胞基因组进行打靶检测,检测结果见图3。由图3可知,与对照细胞相比较,PGRN基因敲除细胞打靶区 TSF的 PCR扩增产物除了有660 bp与野生型大小一致的野生型条带以外,还含有一条较强的470 bp左右的PGRN基因敲除条带。上述结果说明携带 Cas9和 PGRN sgRNA2+sgRNA3的慢病毒已对HEK293 C3-6细胞PGRN基因进行有效的基因打靶敲除。
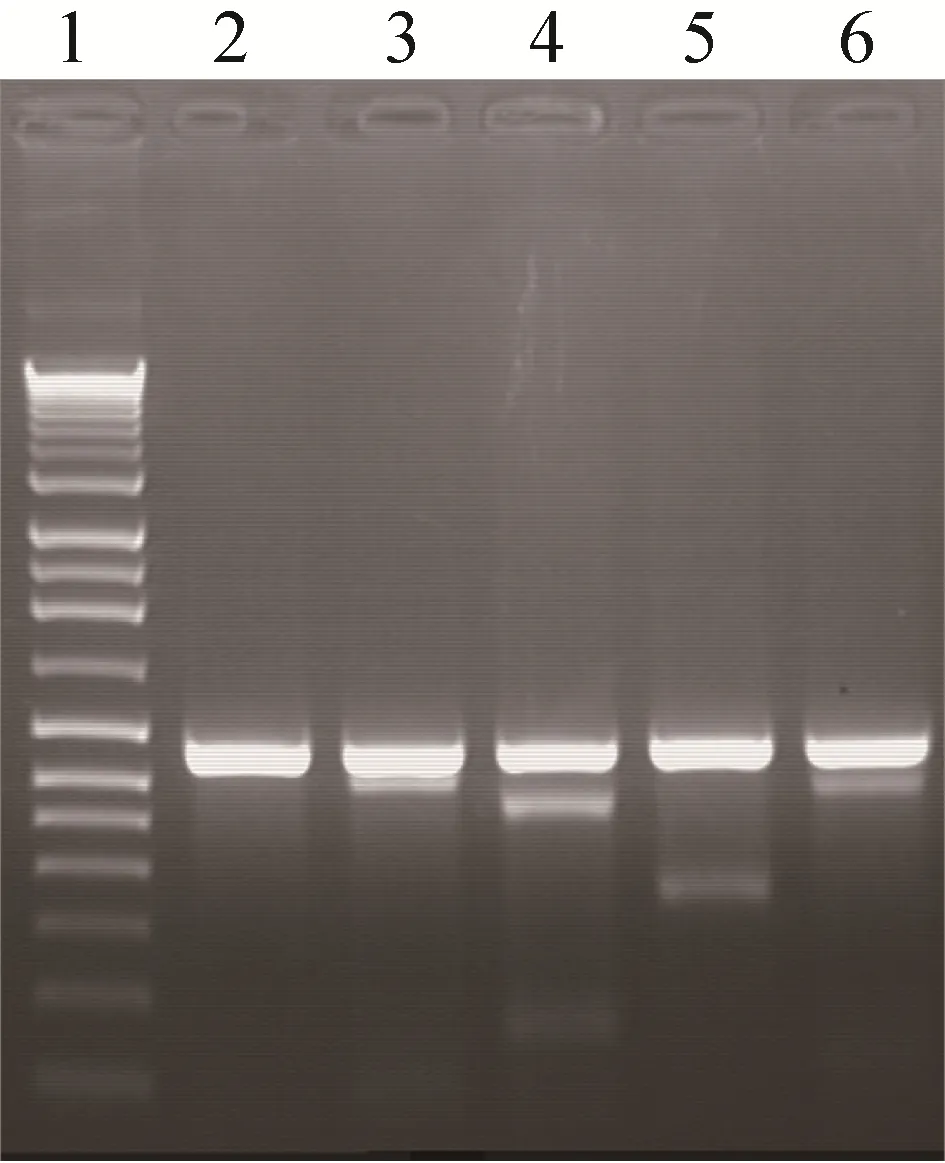
图2 PGRN sgRNA 1–4的生物活性检测Fig.2 Detection the activity of PGRN sgRNA1–4.Lane 1: DNA marker; lane 2: negative control(the product of T7E1 cut the TSF fragment from HEK293 transfected with control vector); lane 3–6: the product of T7E1 cut the TSF fragment from HEK293 transfected with PGRN TSF fragment from HEK293 transfected.
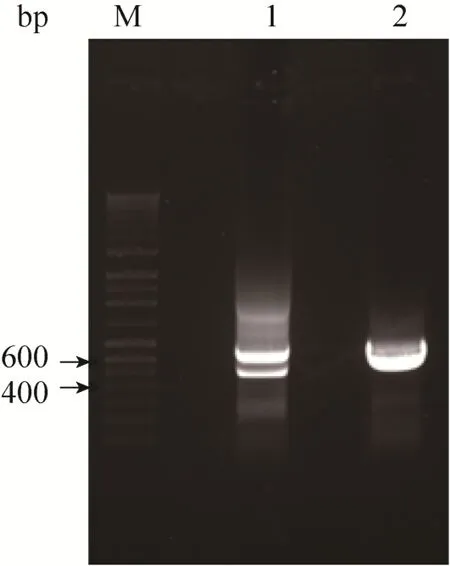
图3 PGRN基因敲除打靶检测Fig.3 Detection the knockout of targeting PGRN gene.Lane M: DNA marker; lane 1: the PCR product of PGRN gene knockout TSF in HEK293 C3-6 cell; lane 2: the PCR product of TSF in control cell.
将上述PGRN基因打靶检测TSF区PCR扩增产物660 bp 和470 bp DNA片段,凝胶回收后送测序,测序结果发现野生型660 bp条带和基因敲除470 bp条带的DNA片段均存在套峰。这说明通过慢病毒打靶敲除后PGRN基因TSF区DNA序列存在有多种基因型(打靶敲除后筛选的HEK293 C3-6细胞不纯),因此对该不纯的HEK293 C3-6细胞进行克隆化筛选。通过克隆化处理,筛选到了 35株阳性单克隆细胞株。提取35株阳性单克隆细胞的基因组,用外、内巢打靶检测引物P1/P2、P3/P4,PCR扩增阳性单克隆细胞 PGRN TSF区基因片段,与野生型 TSF序列相比较,发现 35株阳性克隆细胞均有打靶效果。
选取阳性克隆细胞中状态较好的 13株细胞分析发现,这13株阳性克隆细胞既存在与野生型TSF区大小一致660 bp的DNA条带,又存在400 bp左右的基因敲除DNA条带(图4)。并将13株阳性单克隆细胞的PGRN基因打靶区TSF PCR扩增产物660 bp DNA片段和400 bp左右的DNA片段凝胶回收、连接pGEM-T载体,提取阳性质粒送测序。测序分析结果发现,HEK293 C3-6/C23号阳性细胞为单克隆细胞系,且该细胞的PGRN基因一条链在PGRN基因打靶区 TSF中的 PGRN sgRNA2与sgRNA3之间缺失187 bp;另一条链在PGRN基因打靶区TSF中的PGRN sgRNA2处缺失7 bp(具体见图5)。其他12株阳性单克隆细胞均为一条链为野生型,另一条链在PGRN基因sgRNA2处或sgRNA2和sgRNA3处缺失8-190 bp碱基不等的单倍体基因缺失突变型(具体分析略)。由上述鉴定结果可知,经携带Cas9和PGRN sgRNA2+sgRNA3的慢病毒感染 HEK293 C3-6(Rev-erbβ-/-)细胞进行PGRN基因打靶敲除,再经克隆化筛选成功获得一株PGRN和Rev-erbβ两个基因都完全敲除的 HEK293 C3-6/C23细胞系。
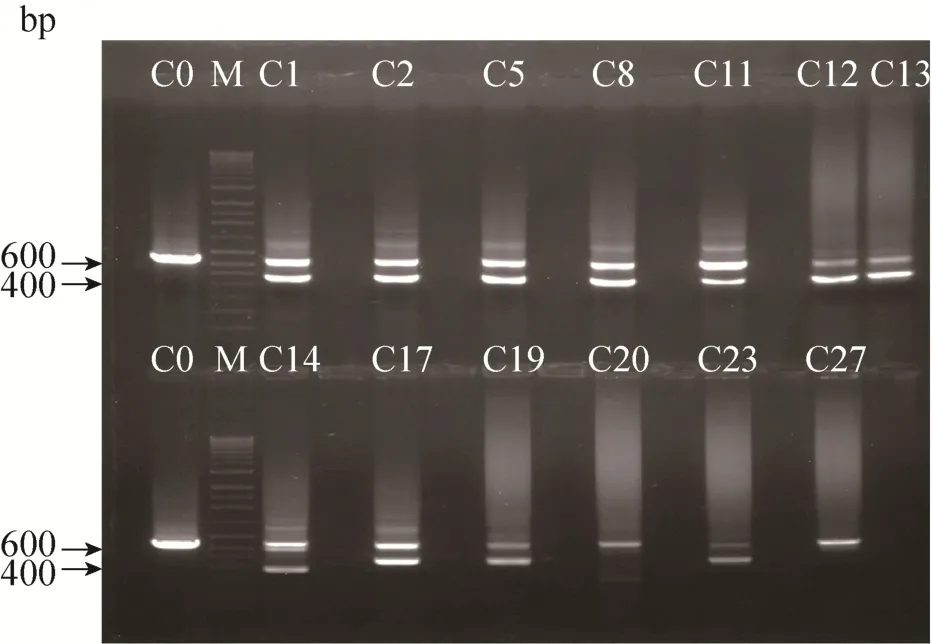
图4 PCR鉴定PGRN基因打靶后筛选的阳性克隆Fig.4 PCR identification genotype of positive clone with PGRN gene knockout.Lane C0: PCR amplification the TSF of control HEK293 C3-6 cells; lane M:DNA marker; lane C1–C27: PCR amplification the TSF of the positive clones of the HEK293 C3-6/C1,C2,C5,C8,C11,C12,C13,C14,C17,C19,C20,C23,C27 cell lines with PGRN gene knockout.

图5 HEK293 C3-6/C23基因敲除细胞系PGRN基因突变型分析Fig.5 Analysis of knockout PGRN gene mutants of HEK293 C3-6/C23.(A) The PGRN target sequence fragment(from PGRN sgRNA2 to sgRNA3) of wild-type.(B) The deletion mutant of PGRN target sequence fragment.One deletion mutant chain lacks of 187 bp locating between sgRNA2 and sgRNA3 gene targeting area(the 187 bp specific deletion base sequences are highlighted on the underlines of wild-type).And the other deletion mutation chain lacks of 7 bp in PGRN sgRNA2 gene targeting area(the 7 bp specific deletion base sequence are showed in green rectangle of wild-type).
2.3 PGRN mRNA和蛋白质在 HEK293 C3-6/C23细胞系中的表达水平
为了验证PGRN基因敲除后是否对HEK293 C3-6/C23细胞系的PGRN mRNA、蛋白质表达水平有影响,采用qRT-PCR和Western blotting法分别检测在 HEK293 C3-6/C23细胞系和对照HEK293 C3-6细胞系中的PGRN mRNA、蛋白质表达水平。qRT-PCR检测结果表明,与对照细胞系相比,PGRN敲除细胞中的PGRN mRNA表达水平呈现极显著的降低(图 6A);同样 Western blotting检测结果显示,对照细胞系中有明显的PGRN蛋白质印迹条带,而在PGRN敲除细胞系HEK293 C3-6/C23中未检测到PGRN蛋白质的印迹条带(图6B)。上述结果一致表明PGRN mRNA和蛋白质表达水平在PGRN敲除细胞系中呈极显著的下降趋势。因此进一步证实,成功构建并获得PGRN和Rev-erbβ双基因敲除的 HEK293 C3-6/C23细胞系。

图6 RT-qPCR和Western blotting检测不同细胞系PGRN mRNA、蛋白质的表达水平Fig.6 RT-qPCR and Western blotting detection the PGRN mRNA and protein expression in different HEK293 cell lines.(A) The PGRN mRNA expression in PGRN knockout cell lines and control cells.1: HEK293 C3-6 control cell lines; 2: the PGRN knockout cell lines of HEK293 C3-6/C23,bP<0.01vs control.(B) The PGRN protein expression level in PGRN knockout cell lines and control cells.1: the cells lysate of HEK293 C3-6 control cell lines; 2: the cells lysate of HEK293 C3-6/C23 cell lines.The anti-PGRN antibody(1:1 000) was used to detect PGRN protein expression in different cell lines,and the anti-GAPDH is internal control.
2.4 PGRN介导 Rev-erbβ对靶基因启动子转录调控的影响
将Rev-erbβ靶基因启动子荧光素酶报告基因表达载体pGL3-hApoCIII promoter-Luci和pGL3-hSrebp-1c promoter-Luci分别同PGRN、Rev-erbβ表达载体等共转染到PGRN和Rev- erbβ双基因敲除HEK293 C3-6/C23细胞系中,转染后48 h收细胞总蛋白,用全波长多功能酶标仪检测各实验组荧光素酶的相对活性见图 7。由检测结果可知,在HEK293 C3-6/C23细胞中回补PGRN不仅可以增强Rev-erbβ对靶基因ApoCIII启动子转录的抑制作用(图 7A),还可以增强 Rev-erbβ对靶基因Srebp-1c启动子转录的激活作用(图7B)。这些结果说明,在HEK293 C3-6/C23细胞内,PGRN与Rev-erbβ相互作用可以激活Rev-erbβ对靶基因启动子转录的调控。
3 讨论
基因组编辑技术已被广泛有效地应用于构建细胞和动物疾病模型、培育生物新品种以及遗传性疾病的治疗研究上。人工核酸酶的出现极大地提高了基因组编辑的效率,彻底改变了该项技术的应用现状,其中CRISPR/Cas9基因组编辑技术操作简单、省时省力、成本廉价等优点使它一出现就很快成为人们热捧的基因编辑技术。
截止目前,已有很多研究团队使用CRISPR/Cas系统成功开展细胞或动物基因编辑及疾病治疗相关研究。虽然与 ZFN和 TALEN技术相比,CRISPR/Cas9系统只需将设计好的20 bp左右的sgRNA和重组的 Cas9蛋白通过一定的方法导入细胞中就可以对细胞特定的基因进行靶向编辑修饰了,但是该技术自最初提出以来一直处于不断改进和发展过程中的[36-39]。自2013年以来,本实验室一直致力于利用CRISPR/Cas9系统开展基因组编辑相关的工作(人源基因敲除细胞系、小鼠疾病模型构建等)[15,34,40]。前期基因打靶工作主要借鉴Cho等[41]提出的提高sgRNA与Cas9系统特异性和减少脱靶效应的策略,通过脂质体共转染打靶载体(含特异的有活性的 sgRNA和重组Cas9) 和供体载体(携带靶基因上、下游同源臂)到靶细胞内进行基因的敲除或敲入等编辑修饰操作。通过这种基因打靶操作方式,一方面比较容易将正常基因或外源标记基因如 eGFP导入靶细胞进行基因纠正或筛选基因敲除细胞系;另一方面,当sgRNA引导Cas9对靶基因DNA 进行特异切割时,导入的同源 DNA模板将会大大提高基因打靶同源重组修复的效率,使切割的DNA发生同源重组,并将外源DNA片段整合到靶基因组特定位点上,从而实现基因的敲入、敲除突变。但是后来研究发现,上述基因打靶操作方式存在许多问题,首先是脂质体转染哺乳动物细胞,特别是一些肿瘤细胞系效率很低,使得最终的打靶效率不高;其次是这种基因打靶方式容易获得一条链为整合型突变,另一条链可能为野生型、缺失突变或整合型突变,三者各占 1/3的几率。因此,要想获得两条 DNA链都发生突变的细胞系概率非常小,往往需要通过增加基因打靶、筛选及克隆化等操作的工作量来实现。鉴于实验室前期基因打靶方式存在诸多问题以及构建PGRN和 Rev-erbβ双基因敲除细胞系的实验目的,本实验重新设计构建了同时携带 Cas9和PGRN双sgRNA的慢病毒打靶载体。利用慢病毒打靶载体对HEK293 C3-6细胞系的PGRN基因进行打靶敲除,药物筛选细胞稳定后,再用含 Cre的慢病毒 pLenti/CMV-Cre-Hygromycin感染有打靶效果的细胞(以除去整合到细胞基因组中的Loxp-Cas9-sgRNA2-U6-sgRNA3-U6-Loxp),通过克隆化、基因测序鉴定等操作最终获得了PGRN和Rev-erbβ双基因敲除 HEK293 C3-6/C23细胞系。利用改进的慢病毒打靶载体进行基因打靶操作方式,直接用携带Cas9和PGRN双sgRNA的慢病毒感染细胞进行基因打靶,在打靶的同时由于没有引入同源DNA片段,故切割的DNA主要通过非同源末端连接方式进行 DNA修复,会在切割位点上引起插入或缺失突变,从而引发目的基因编码蛋白质发生移码突变以达到目的基因敲除的目的。这种利用改良的慢病毒打靶载体进行基因打靶操作方式,不仅通过提高打靶载体导入靶细胞的效率来提高打靶效率;而且可以缩短和简化基因打靶、筛选及克隆化等操作的周期和过程。
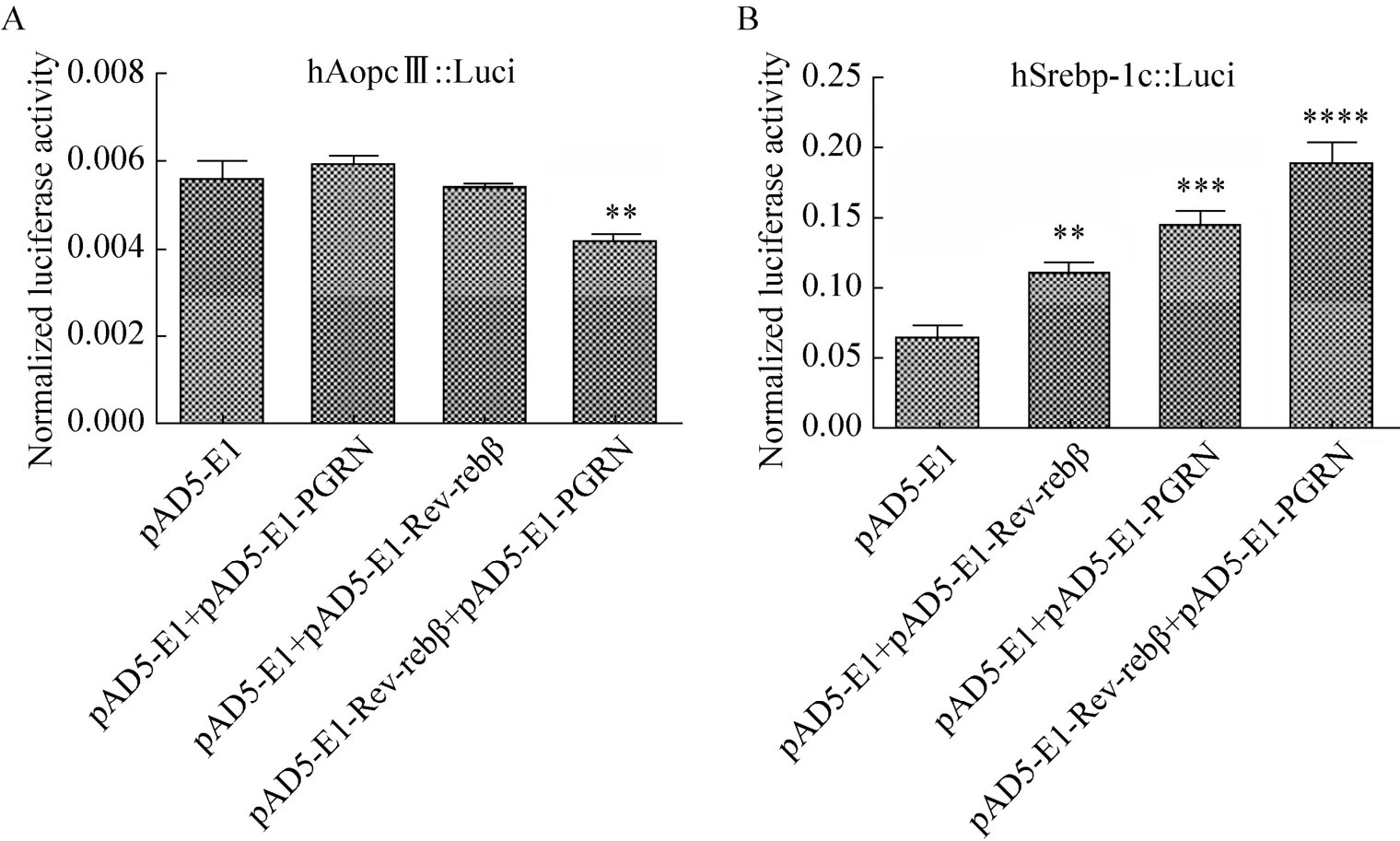
图7 PGRN介导Rev-erbβ对靶基因启动子活性调控的影响Fig.7 Effect of PGRN mediating Rev-erbβ on the target gene promoter activity.(A) Effect of PGRN mediating Rev-erbβ on the hApoCIII promoter activity in HEK293 C3-6/C23 cells.B: Effect of PGRN mediating Rev-erbβ on the hSrebp-1c promoter activity in HEK293 C3-6/C23 cells.pAD5-E1 as control(150 ng pGL3-hApoCIII/hSrebp-1c promoter-Luci plasmid,200 ng pAD5-E1 plasmid and 20 ng pRL-CMV-Renilla-Luci plasmid); pAD5-E1+pAD5-E1-PGRN(150 ng pGL3-hApoCIII/hSrebp-1c promoter-Luci plasmid,100 ng pAD5-E1 plasmid,100 ng pAD5-E1-PGRN plasmid and 20 ng pRL-CMV-Renilla-Luci plasmid); pAD5-E1+pAD5-E1-Rev-erbβ(150 ng pGL3-hApoCIII/hSrebp-1c promoter-Luci plasmid,100 ng pAD5-E1 plasmid,100 ng pAD5-E1-Rev-erbβ plasmid and 20 ng pRL-CMV-Renilla-Luci plasmid); pAD5-E1-PGRN+pAD5-E1-Rev-erbβ(150 ng pGL3-hApoCIII/hSrebp-1c promoter-Luci plasmid,100 ng pAD5-PGRN plasmid,100 ng pAD5-E1-Rev-erbβ plasmid and 20 ng pRL-CMV-Renilla-Luci plasmid).** P<0.01,*** P<0.005,****P<0.001 vs control.
本研究在前期构建携带外源基因 eGFP的Rev-erbβ基因敲除细胞系(HEK293 C3-6) 的基础上,利用携带重组 Cas9和串联较高活性的PGRN sgRNA2和sgRNA3的慢病毒感染HEK293 C3-6细胞对PGRN基因进行打靶敲除,再经药物筛选、克隆化和基因测序等操作,成功获得一株PGRN和Rev-erbβ双基因敲除的 HEK293 C3-6/C23细胞系。所构建的PGRN和Rev-erbβ双基因敲除HEK293 C3-6/C23细胞系,彻底解决了传统 RNAi技术的不彻底性和瞬时性对基因功能研究的不足。随后,利用所构建的 HEK293 C3-6/C23细胞系进行研究表明PGRN与Rev-erbβ相互作用,前者可以影响Rev-erbβ对靶基因启动子转录的调控。总之,这些研究为阐明PGRN介导 Rev-erbβ对靶基因转录调控的分子机制和生理意义奠定了坚实的基础和提供了有效的研究工具。
致 谢特别感谢陕西师范大学基因治疗室李雅博士提供的慢病毒打靶载体,张伟锋博士对基因打靶提供的技术支持。
猜你喜欢
环球时报(2022-09-20)2022-09-20 15:18:57
今日农业(2021年11期)2021-08-13 08:53:24
今日农业(2020年24期)2020-12-15 16:16:00
兽医导刊(2016年12期)2016-05-17 03:51:50
山东医药(2015年14期)2016-01-12 00:39:43
江苏大学学报(医学版)(2015年2期)2015-04-17 06:49:51
中国医药导报(2015年26期)2015-02-28 22:07:44
现代检验医学杂志(2015年4期)2015-02-06 02:02:06
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
