
复方氨酚苯海拉明片的溶出曲线质量评价
2018-10-15 07:25陈星宇刘美玲朱恒怡徐小平钱广生
中国测试 2018年8期
陈星宇,刘美玲,朱恒怡,袁 军,徐小平,钱广生
(1.四川大学华西药学院,四川 成都 610041;2.四川省食品药品检验检测院,四川 成都 611731)
0 引 言
复方氨酚苯海拉明片是由对乙酰氨基酚(BCS4类)、咖啡因(BCS3类)、盐酸苯海拉明(BCS1类)和盐酸麻黄碱(BCS3类)4种成分组成的复方制剂,共两个规格,规格一中每片含对乙酰氨基酚300 mg、咖啡因30 mg、盐酸麻黄碱7.5 mg、盐酸苯海拉明7.5 mg,规格二中各主成分量为规格一含量的一半。该制剂具有良好的解热镇痛效果。临床常用于治疗伤风感冒引起的发热、头痛,且伴有咳喘症状者[1-2]。该品种的法定检验标准《国家药品标准》(化学药品地方标准上升国家标准)第十二册[WS-10001-(HD-1152)-2002]中未收载溶出度检查方法,且未检索到该品种溶出曲线研究的文献报道。体外溶出度试验现已作为评价固体制剂内在品质的重要手段之一[3-4]。对不同厂家同一剂型溶出度的全面深入研究以及溶出曲线的准确客观绘制,可揭示各制剂工艺的差别和内在品质的优劣,也可初步为体内生物利用率和生物等效性提供参考[5]。为了从体外试验探究该制剂的临床疗效,本研究对7个企业生产的复方氨酚苯海拉明片中4种主成分的溶出曲线同时进行检测并比较,对不同厂家药品的内在质量进行评价,对影响溶出行为的处方、环境、包材因素进行研究分析,为临床用药和药品质量稳定提供参考。
1 仪器与试药
1.1 仪 器
Agilent Technologies 708-DS Dissolution Apparatus,配 8000 Dissolution Sampling Station(美 国Agilent公司);Waters Acquity UPLC色谱仪(美国Waters公司);ZKT-18F真空脱气仪(天大天发科技有限公司);Sartorius CP225D电子天平 (德国Sartorius公司);SevenExcellece pH 计(瑞士 Mettler-Toledo 公司);超纯水机(ULUP-IV-20T)。
1.2 试 剂
对乙酰氨基酚(中国食品药品检定研究院,批号:100018-201409,含量:99.9%),咖啡因(中国食品药品检定研究院,批号:171215-201512,含量:99.9%),盐酸麻黄碱(中国食品药品检定研究院,批号:171241-201007,含量:99.7%),盐酸苯海拉明(中国食品药品检定研究院,批号:100066-200807,含量:99.9%);复方氨酚苯海拉明片,规格一:每片含对乙酰氨基酚300 mg、咖啡因30 mg、盐酸麻黄碱7.5 mg、盐酸苯海拉明7.5 mg(A厂,黑龙江省,批号:20161124;B厂,吉林省,批号:20161103;C厂,吉林省,批号:20160901;D厂,吉林省,批号:20160901;E厂,吉林省,批号:20170109;F 厂,黑龙江省,批号:20170201),规格二:每片含对乙酰氨基酚150 mg、咖啡因15 mg、盐酸麻黄碱3.75 mg、盐酸苯海拉明3.75 mg(G厂,吉林省,批号:03-160909);参照制剂酚麻美敏片(H 厂,上海市,批号:151015726)、乙腈、磷酸为色谱纯,氯化钠、盐酸、冰醋酸、乙酸钠、磷酸氢二钠、磷酸二氢钾均为分析纯。
2 检测方法
2.1 溶出方法的确定
2.1.1 溶出介质的选择
选择4种不同pH值的溶出介质来模拟消化道内的体液[6](pH1.2、pH4.0、pH6.8 的缓冲液及水),配制方法为:1)pH 1.2盐酸溶液:称取氯化钠2.0 g,加水适量溶解,加盐酸7 mL,再加水稀释至1 000 mL,即得;2)pH4.0醋酸盐缓冲液:称取三水合醋酸钠1.22 g,加入冰醋酸溶液2.337 mL,加水溶解并稀释至1 000 mL,即得;3)pH6.8磷酸盐缓冲液:称取磷酸二氢钾3.4 g和无水磷酸氢二钠3.55 g,加水适量溶解并定容至1 000 mL,再稀释一倍,即得;4)水:纯化水。上述介质在使用前均采用真空脱气处理。
2.1.2 溶出方法的选择
最常用的溶出方法为篮法或桨法,对于片剂的溶出考察,大多采用桨法[7],本方法采用桨法。
2.1.3 溶出参数的选择
转速对制剂的溶出速率影响明显[8],通过预实验发现50 r/min较75 r/min更有区分力,因此选择50 r/min;人体消化道内体液总体积接近于900 mL[9],因此溶出介质体积采用900 mL,为兼顾检测方法,处方规格二采用500 mL溶出介质。
2.1.4 取样点的选择
基于预实验结果,选择各溶出介质中的取样点分别为:1)pH1.2盐酸溶液:5,10,20,30,45,60,90,120 min;2)pH4.0的醋酸盐缓冲液:5,10,20,30,45,60,90,120 min;3)pH6.8 的磷酸盐缓冲盐:5,10,20,30,45,60,90,120 min;4)水:5,10,20,30,45,60,90,120 min。
2.1.5 参照制剂的选择
未查询到国外复方氨酚苯海拉明片上市制剂。因此选择对乙酰氨基酚处方量相似的酚麻美敏片(规格:乙酰氨基酚325 mg,盐酸伪麻黄碱30 mg,氢溴酸右美沙芬10 mg,马来酸氯苯那敏2 mg)作为参照制剂进行溶出曲线研究。
2.2 测定方法的建立和验证
对于复方制剂,首选液相方法同时对几个有效成分进行溶出量的测定。本文建立了UPLC检测方法,实现了快速分析测定4个主成分。
2.2.1 色谱条件
色谱柱:Waters ACQUITY UPLC BEH C18柱(2.1 mm×50 mm,1.7 μm);流动相:水(A)(磷酸调pH 至2.5)-乙腈(B),梯度洗脱(0~1 min,5%B;1~2 min,5%~70%B;2~2.5 min,70%~5%B;2.5~3 min,5%B);流量0.5 mL/min;检测波长分别为216 nm(对乙酰氨基酚和盐酸苯海拉明)和205 nm(咖啡因和盐酸麻黄碱);进样量分别为2 μL(对乙酰氨基酚和咖啡因)和20 μL(盐酸麻黄碱和盐酸苯海拉明);柱温为30℃。该色谱条件下各成分的分离度良好,如图1所示。
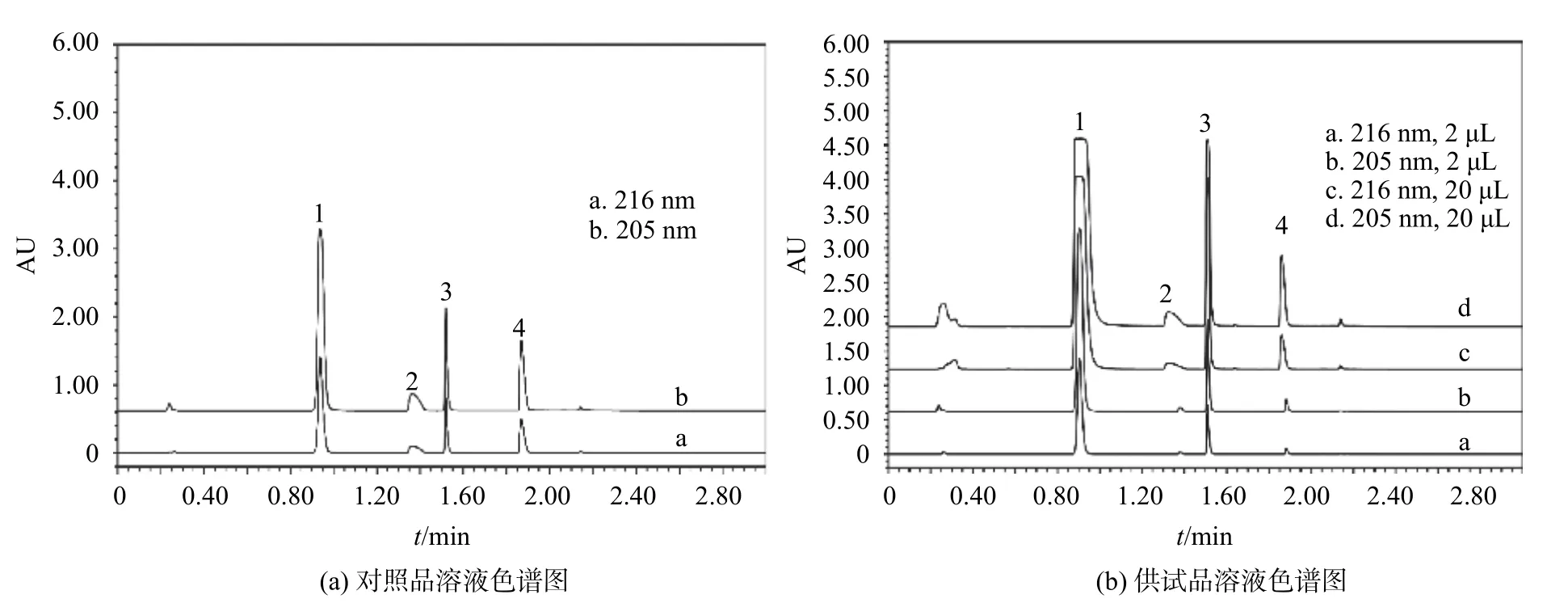
图1 复方氨酚苯海拉明片中4种成分的UPLC分离色谱图
2.2.2 对照品溶液的制备
分别精密称取对乙酰氨基酚、咖啡因、盐酸麻黄碱、盐酸苯海拉明对照品适量,用溶出介质溶解并稀释制成含上述各成分约0.33 mg/mL,0.033 mg/mL,0.083 mg/mL,0.083 mg/mL的混合对照品溶液。
2.2.3 供试品溶液的制备
取各时间点下的溶出液用0.45 μm的滤膜过滤,弃去3mL初滤液后,取续滤液即得。
2.2.4 方法学验证
1)专属性试验
分别取空白溶剂、空白辅料、对照品溶液和供试品溶液各20 μL,按2.2.1项下色谱条件进样测定,结果如图2所示,表明各介质及空白辅料均不干扰主成分的检出,专属性强。
2)滤膜吸附试验
于同一溶出杯中取两份样品,一份溶液用0.45 μm微孔滤膜依次弃去初滤液 0,1,2,3,4,5,6,7 mL,取续滤液进样测定[10]。另一份溶液离心(10 000 r/min,10 min)后取上清液进样测定。结果表明,弃去3 mL初滤液后,滤膜的吸附率均小于2.0%,对各有效成分的测定无影响。
3)线性范围
分别精密称取对乙酰氨基酚对照品33.33 mg置于10 mL量瓶中,咖啡因对照品16.65 mg置于50 mL量瓶中,盐酸麻黄碱对照品20.75 mg置于25 mL量瓶中,盐酸苯海拉明对照品20.75 mg置于25 mL量瓶中,加入2.1.1项下的4种溶出介质使溶解并稀释至刻度,摇匀,作为混合对照品贮备液;分别精密量取对照品贮备液适量,用相应的溶出介质稀释制成5%、10%、20%、50%、70%、100%、120%系列浓度的混合对照品溶液,照2.2.1项下色谱条件进样检测,以4种主药成分的浓度(mg/mL)对峰面积进行最小二乘法线性回归拟合。结果表明,在4种溶出介质中,对乙酰氨基酚、咖啡因、盐酸麻黄碱、盐酸苯海拉明分别在 16.50~400.00 μg/mL、1.65~40.00 μg/ml、4.15~99.60 μg/mL 和4.15~99.60 μg/mL浓度范围内与峰面积线性关系良好。
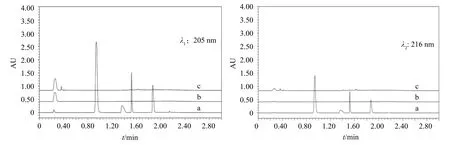
图2 专属性试验色谱图(a.对照品溶液;b.溶剂;c.空白辅料)
4)精密度试验
分别取4种溶出介质中对乙酰氨基酚、咖啡因、盐酸麻黄碱、盐酸苯海拉明浓度分别为0.33 mg/mL,0.033 mg/mL,0.083 mg/mL,0.083 mg/mL的混合对照品溶液,照2.2.1项下色谱条件连续进样6次,记录峰面积,计算RSD值。结果4种溶出介质中各主药成分峰面积的RSD值均小于2.0%,表明本方法精密度良好。
5)回收率试验
按处方量比例的5%、50%、120%精密称取4种主药成分对照品各3份,加入处方量的辅料,加4种溶出介质配制成低、中、高浓度的回收率试验溶液,照2.2.1项下色谱条件进样检测,记录峰面积。4种主成分在pH1.2、pH4.0、pH6.8介质中回收率均在95.0%~105%范围内。在介质水中,含羧甲基淀粉钠辅料的厂家,盐酸麻黄碱和盐酸苯海拉明的回收率只有72.2%和62.5%(远小于95%),所以,介质水中只考察了不含羧甲基淀粉钠辅料的厂家样品的溶出曲线。
6)重复性试验
精密称取复方氨酚苯海拉明片粉末6份,加4种溶出介质溶解,过滤,取续滤液进样检测,记录峰面积。结果4种主成分含量RSD值均小于2.0%,表明方法的重复性良好。
7)稳定性试验
取2.2.2项下对照品溶液和2.2.3项下供试品溶液,分别于 0,2,4,6,8,12,18 h按 2.2.1 项下色谱条件测定,结果在18 h内4种主成分在4种溶出介质中峰面积的RSD均小于2%,表明对照品溶液和供试品溶液在18 h内稳定。
3 溶出曲线测定结果
按照第2节中确定的溶出曲线检测方法,检测了参照制剂和7个厂家生产的复方氨酚苯海拉明片的溶出曲线。现将检测结果汇总如图3、图4所示(因数据较多,此处仅列出BCS分类4类且所占比重最大的对乙酰氨基酚的典型溶出曲线)。
3.1 各企业样品在不同介质中溶出曲线的典型图谱(溶出量为6片的均值)
从图3可以看出,在4种介质中参照制剂中对乙酰氨基酚的溶出行为相似,溶出速率快,批内差异小。而厂家E的样品溶出行为差异较大,溶出速率慢。由于复方氨酚苯海拉明片各生产企业的样品中4种主成分在pH1.2、pH4.0、pH6.8、水中6片样品溶出量RSD值不满足“第一个时间点组内RSD小于20%,以后时间点组内均小于10%”的要求[11],批内差异大,所以不再进行批间溶出行为比较。
3.2 相同溶出介质中各企业样品溶出曲线比较
由图4可以看出,各生产企业的复方氨酚苯海拉明片在相同介质中的溶出行为差异很大,如在pH1.2介质中B厂家30 min达到完全溶出,而E厂家120 min才能达到完全溶出。提示各生产企业的样品在人体内吸收可能会有差异,疗效也不同,不同生产企业的产品效果不能保证一致。
4 影响溶出行为的因素分析
针对复方氨酚苯海拉明片批内溶出速率差异大,溶出较参照制剂缓慢,各生产企业样品之间溶出行为差异大的现状,本研究对影响本品溶出行为的因素进行了考察。
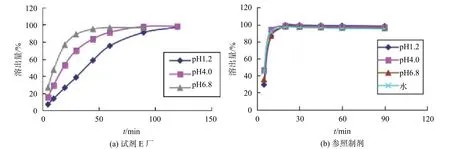
图3 不同介质中对乙酰氨基酚的溶出曲线
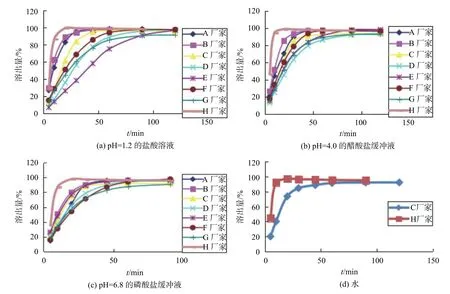
图4 相同介质中各企业样品中对乙酰氨基酚的溶出曲线
4.1 高湿影响因素考察
选择区分力较强的pH1.2溶出介质,考察样品(G厂家)在高湿(RH90%,25 ℃)条件下,不同包装程度放置一段时间后,样品中各主成分溶出行为的变化情况如图5所示。结果表明,水分对样品溶出有一定的影响,会使4个主成分的溶出速率和程度变缓,溶出行为差异变大。
4.2 片剂硬度考察
采用YPD-300C型智能片剂硬度测定仪对样品硬度进行了测定,考察产品工艺稳定性和与样品溶出速率的关系。结果表明,各厂家生产的样品硬度从12.56~130.22 N不等,差异较大,其中D厂家批间差异大(52.11~83.73 N),其可能也是导致该企业样品溶出行为差异较大的原因。
4.3 制剂处方考察
经调研,7个生产厂家所使用的辅料种类相近,都使用了淀粉、蔗糖、硬脂酸镁,仅在崩解剂的使用上存在差异。D、E、B、F 4个厂家都使用了羧甲基淀粉钠,另外F厂家还使用了羟丙甲纤维素,A厂家使用了低取代羟丙纤维素,C厂家使用了预胶化淀粉,G厂家未使用任何崩解剂,该厂家样品的溶出速率相比其他厂家较为缓慢。
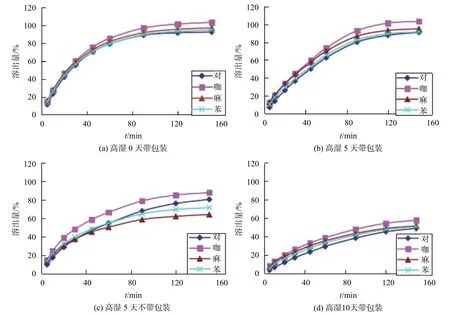
图5 高湿条件下市售厂家G溶出曲线变化趋势图
5 结束语
本文对复方氨酚苯海拉明中片中的4种成分进行UPLC 检测,通过对复方氨酚苯海拉明片溶出方法、检测方法、溶出曲线的考察,该方法的精密度和重复性的相对标准偏差均小于2.0%,对乙酰基氨酚、咖啡因、盐酸麻黄碱和盐酸苯海拉明的分离度和线性关系良好。回收率实验中,含羧甲基淀粉钠辅料的厂家,盐酸麻黄碱和盐酸苯海拉明在水中的回收率较低,说明了介质水不适用于含羧甲基淀粉钠样品溶出行为的考察,同时药品片与片之间在体内的吸收速度和程度也会存在差异,高湿实验也揭示了生产企业应关注药品包装材料的质量,从而确保药品的稳定性。
猜你喜欢
药学实践杂志(2022年3期)2022-05-27
药学实践杂志(2021年6期)2021-12-04
山西化工(2021年5期)2021-01-25
保健与生活(2020年4期)2020-03-02
中国医院用药评价与分析(2018年5期)2018-06-19
天然产物研究与开发(2018年5期)2018-06-13
中国药物经济学(2015年1期)2015-12-14
家庭医学(2014年6期)2014-09-11
中国药业(2014年17期)2014-05-26
中国药业(2014年24期)2014-05-26
