
17α-羟化酶缺陷症合并男性女性化的影像学特征
2018-10-08 01:28王勇田小丽孙浩然
放射学实践 2018年9期
王勇, 田小丽, 孙浩然
先天性肾上腺皮质增生(congenital adrenal hyperplasia,CAH)是指肾上腺在皮质醇的合成途径中必需酶的遗传性缺陷所导致的一组疾病[1],其中17α-羟化酶缺陷症是CAH的一种很少见的类型,仅占CAH的1%[2]。与其他类型CAH不同,17α-羟化酶缺陷症以性发育障碍为主要表现,患者常因原发性闭经或青春期延迟而就诊。对于染色体46XX者,17α-羟化酶缺乏时雌激素和睾酮等性腺激素的合成产生障碍,导致女性第二性征不发育和原发性闭经;而染色体46XY者在胚胎发育期睾丸发育停留在早期状态,不能形成阴茎及阴囊,外生殖器呈女性化表现[3]。17α-羟化酶缺陷症所致男性女性化的异常表现隐蔽,多数患者于青春期后就诊且容易误诊。影像学检查对本病有很高的提示诊断价值,并对选择治疗措施有很大帮助。本文回顾性分析9例17α-羟化酶缺陷症患者的临床及影像学资料,总结其影像学表现特征,旨在提高对本病的认识。
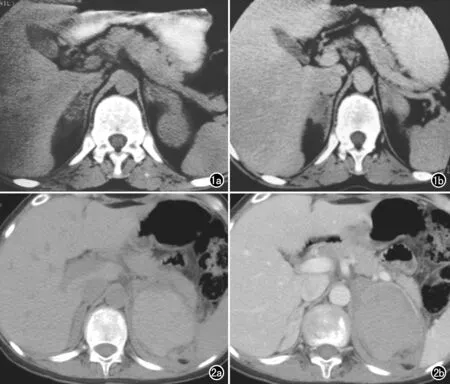
图1 17α-羟化酶缺陷症患者。a) CT平扫示双侧肾上腺各肢明显均匀增粗; b) CT增强扫描示双侧肾上腺均匀增粗并明显强化。 图2 17α-羟化酶缺陷症患者左侧肾上腺腺瘤自发破裂。a) CT平扫示右肾上腺弥漫增粗,左肾上腺可见中央高密度、边缘稍低密度的不均质肿物,大小约为8.5 cm×7.0 cm×5.0 cm; b) CT增强扫描示右肾上腺明显均匀强化,左肾上腺可见肿块高密度区无强化,周围呈轻度不均匀强化。
材料与方法
搜集1998年5月-2017年9月天津医科大学总医院经临床生化检验和遗传学证实的9例17α-羟化酶缺陷症合并男性假两性畸形患者,所有患者根据临床体格检查、激素检查结果、影像学检查结果和染色体核型分析结果做出诊断。9例患者社会性别均为女性,年龄分别为9、11、13、14、16、17、17、20和26岁。其中,9岁和11岁患者表现为高血压;13岁、14岁、16岁和17岁患者因原发闭经就诊;17岁和20岁患者为姐妹,姐姐因高血压就诊,妹妹系家族内筛查发现;26岁患者因左侧腰痛发现肾上腺肿物就诊。所有患者均进行双侧肾上腺、盆腔下腹部CT和/或MRI检查,16岁及以上的5例患者另行左手X线骨龄测定。所有患者均进行了外周染色体核型分析。
结 果
1.临床表现与实验室检查结果
所有患者外阴均为幼女型,乳腺未发育,均无腋毛和阴毛生长。年龄14岁以上的6例患者喉结不大,四肢细长,手足较大。9例患者均进行了肾上腺激素水平测定,血促肾上腺皮质激素(Adrenocorticotropic Hormone,ACTH)水平均明显高于正常范围,治疗前血钾水平不同程度低于正常,血尿皮质醇水平正常或轻度增高,所有患者血尿醛固酮水平均有不同程度增高,血浆肾素活性不同程度减低。6例14岁以上患者检测了性激素水平,其中4例睾酮水平明显低于正常男性水平,1例高于正常,1例接近正常;4例患者雌二醇水平低于正常女性,2例位于女性正常范围。所有患者染色体核型分析结果均为 46XY。
2.影像学表现
双侧肾上腺显著弥漫增粗合并结节:9例患者均可见双侧肾上腺显著弥漫增粗(图1),其单侧肾上腺横轴面最大面积为342.2~884.6 mm2,平均640.1 mm2。其中1例患者在双侧肾上腺弥漫增粗基础上伴发双侧肾上腺多发结节,增强扫描可见明显均匀强化;另1例患者右肾上腺弥漫增粗,左肾上腺肿物自发破裂出血行肾上腺手术切除,病理诊断为左肾上腺皮质腺瘤伴出血(图2)。
生殖系统异常:9例患者盆腔MRI检查均未见子宫和卵巢,阴道中上段2/3未见显示(图3)。1例11岁患者MRI图像上膀胱下方可见类似前列腺的软组织结构,其余患者未见前列腺和精囊。9例患者均未见男性外生殖器,4例16岁以下患儿和1例17岁患者行腹部超声和MRI检查,双侧腹股沟区和盆腔未见隐睾,17岁、20岁和26岁3例患者发现双侧腹股沟软组织结节,行双侧腹股沟管探查和肿物切除,病理证实为隐睾。
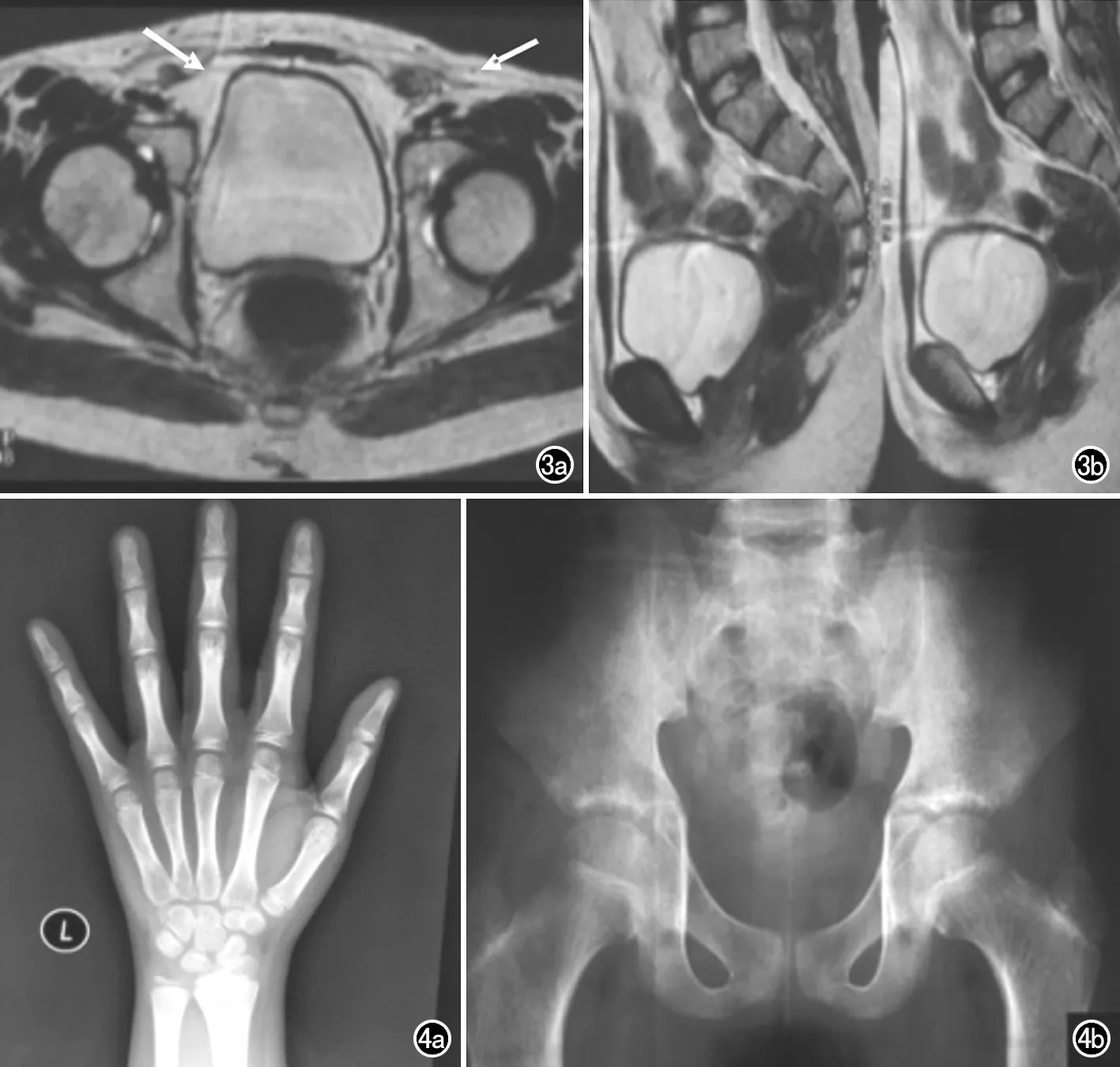
图3 17α-羟化酶缺陷症患者,17岁,盆腔MRI扫描既未见子宫和卵巢,也未见前列腺和精囊,双侧腹股沟管可见软组织信号结节(箭),经手术探查证实为隐睾。a) 横轴面T2WI图像; b) 矢状面T2WI图像。 图4 17α-羟化酶缺陷症患者。a) 左腕部X线正位片示掌指骨骨骺与干骺端未闭合,第1掌骨旁籽骨未出现,腕骨中豆骨未出现; b) 骨盆X线正位片示双侧髋臼和股骨头骨骺未闭合。
骨骺闭合延迟:6例年龄14岁以上的患者行左手X线骨龄检查和骨盆X线检查,均发现骨龄较正常男孩发育迟缓2~4年。患儿左手X线检查均表现为掌指骨骨骺与干骺端未联合,第1掌骨旁籽骨未出现,14岁和17岁患者豆骨未出现(图4)。骨盆X线检查显示双侧髋臼和股骨头骨骺未闭合。
讨 论
17α-羟化酶缺陷所致CAH与绝大多数的其余类型CAH表现不同,性征异常表现隐蔽,多数患者于青春期后就诊且容易误诊。本组9例男性女性化患者均具有典型的17α-羟化酶缺陷症表现,即高血压、低血钾、女性外生殖器、缺乏青春期性腺发育。
CAH中酶缺乏包括很多种类,如21-羟化酶、11β-羟化酶、17α-羟化酶、3β-类固醇脱氢酶、18-羟化酶和胆固醇碳链酶等,以上任何种类的酶缺乏都会导致皮质激素合成障碍,导致垂体代偿性增加ACTH分泌,引起双侧肾上腺皮质增生[4]。CAH症状包括失盐症群、性征异常、高血压伴低血钾、色素沉着等,其中性征异常具有两类表现:21-羟化酶、11β-羟化酶缺陷导致雄激素过多,表现为女性男性化和男性性早熟;雄激素合成障碍由17α-羟化酶、3β-类固醇脱氢酶和胆固醇碳链酶等缺陷造成,表现为男性女性化和女性月经延迟[5,6]。本组患者均为男性17α-羟化酶缺陷症患者,除了具有CAH典型的高血压伴低血钾表现之外,还由于雄激素合成障碍而表现为男性女性化。
自1966年由Biglieri等[3]报道第1例17α-羟化酶缺乏症至今,全世界文献报道逾200例。本组患者ACTH水平增高,血钾水平低于正常,醛固酮水平增高,血浆肾素活性减低,其病理生理基础是由于17α-羟化酶缺乏阻断了糖皮质激素和性激素的合成路径,造成去氧皮质酮大量增加,该物质同时具有糖皮质激素和盐皮质激素的作用,前者足以代偿皮质醇,因此患者均表现为血尿皮质醇水平正常或轻度增高,极少出现肾上腺皮质功能不足,因而临床不易发现;但去氧皮质酮可引起钠潴留、高血压、低血钾和乏力,这些表现是17α-羟化酶缺乏症患者就诊的原因之一,本组9例患者中3例因高血压就诊[7]。由于患者糖皮质激素合成减少, ACTH反应性分泌增加,双侧肾上腺皮质受到持续刺激导致增生[8]。本组患者影像学表现为肾上腺明显弥漫性对称性增粗,肾上腺横轴面的平均最大面积达640.1 mm2,较正常值(150 mm2)增大3倍。本组2例患者合并增生结节或皮质腺瘤,其中1例患者的腺瘤自发破裂。双侧肾上腺的显著增粗对该病的诊断具有提示作用。
17α-羟化酶是去氢表雄酮和雄烯二酮合成中的转化酶,此酶缺乏会导致雌激素和睾酮的合成障碍,造成女性第二性征不发育,表现为性幼稚、原发性闭经,而男性在胚胎发育中缺乏睾酮时,睾丸发育停留在早期阶段,且不能形成阴茎及阴囊,外生殖器呈女性化表现[9]。行影像学检查时子宫、双侧附件结构以及阴道中上段2/3缺如,同时前列腺和精囊缺如的表现具有特征性。本组9例患者中3例发现双侧腹股沟隐睾,在17α-羟化酶缺乏症患者中隐睾的发现除对诊断具有决定性作用以外,还对患者的后续治疗具有指导意义,隐睾一经发现,需手术切除以防恶变。
雌激素具有增加钙盐沉积、加速骨骺闭合的作用。17α-羟化酶缺乏症患者雌激素合成受损,常见骨质疏松、骨骺闭合延迟,相关文献报道患者骨龄可小于实际年龄2~11岁不等[10]。本组6例14岁以上患者按正常男孩骨龄估算,骨龄发育迟缓2~4年,X线表现为骨骺未闭合、腕骨未出齐等,与其他发育障碍或骨代谢疾病不同,患者的骨骺结构无明显异常,身高正常,无其他系统发育障碍,可资鉴别。
肾上腺病变导致的性征异常除CAH外,还可由肾上腺皮质腺瘤和皮质癌引起,后者表现为单侧肾上腺肿块而不合并双侧肾上腺弥漫增粗,因此影像学表现容易分辨。临床症状不明显的双侧肾上腺增粗需与肾上腺结核、淋巴瘤、转移瘤和组织胞浆菌病等鉴别:①肾上腺结核表现为肾上腺弥漫增粗,常伴有钙化,增强扫描表现为边缘强化,中央呈低密度不强化,临床表现为肾上腺皮质功能减低合并皮肤色素沉着[11];②少数肾上腺淋巴瘤可导致皮质功能减低,双侧肾上腺增粗非常明显,双侧肾上腺直径>5 cm对于肾上腺淋巴瘤的诊断具有很大的提示作用[12];③肾上腺转移溜常见于中老年患者,多具有原发肿瘤病史;④组织胞浆菌病常见于细胞免疫缺陷患者[13],常表现为双侧肾上腺增粗,无肾上腺功能减退症状。
综上所述,17α-羟化酶缺陷男性女性化患者发病隐匿,原发性闭经和第二性征不发育是就诊的主要原因,而高血压、低血钾多在患者就医过程中发现,临床表现为高血压、低血钾和/或原发性闭经、青春期延迟而就诊的青年女性,影像学检查对于提示诊断具有很高的价值。双侧肾上腺显著弥漫性增粗伴结节、生殖系统发育异常和骨骺闭合延迟是17α-羟化酶缺陷合并男性女性化的特征性表现。
猜你喜欢
中国实用医药(2022年18期)2022-09-20
中华实用诊断与治疗杂志(2022年2期)2022-09-02
保健与生活(2022年10期)2022-05-06
热带作物学报(2020年9期)2020-10-29
科学24小时(2020年5期)2020-08-02
流行色(2020年9期)2020-07-16
中国现代医生(2020年12期)2020-07-04
心血管外科杂志(电子版)(2018年1期)2018-11-08
文艺生活·中旬刊(2017年11期)2018-01-05
东方教育(2017年14期)2017-09-25
