
Sleeping Beauty转座子介导敲低PEX2转基因载体的构建
2018-09-21 11:49孙潇蔡梦娇张丹韩苏夏
西部医学 2018年9期
孙潇 蔡梦娇 张丹 韩苏夏
(西安交通大学第一附属医院肿瘤放疗科,陕西 西安710061)
转座子(Transposon)是一类可以在基因组中改变自身位置的DNA序列,改变位置的过程被称为转座(transposition)。睡美人(Sleeping Beauty,SB)转座子属于 Tcl/mariner 家族,近几年在哺乳动物中研究较为广泛。SB转座系统包括两端携带反向重复序列(inverted terminal repeat , ITR)的转座子和编码转座酶的序列,可以进行“剪切粘贴”的转座过程,即通过转座酶识别转座子两端的反向重复序列将其剪切,介导转座子整合到基因组中新的位点[1]。SB转座系统的应用为基因研究的体外和体内实验提供了新的基因编辑技术。 Magnani CF等利用SB转座系统将含有CD19CAR的转座子插入到细胞因子诱导型杀伤细胞(cytokine-induced killer cell,CIK cell)的基因组中,形成CARCIK-CD19细胞,并对该细胞治疗急性淋巴细胞白血病进行临床前疗效和安全性评估[2]。Gao B等应用SB转座系统构建骨骼肌细胞特异性高表达mIGF-1的小鼠模型,发现 mIGF-1在小鼠骨骼肌中过度表达可促进肌纤维肥大和肌肉产生,并提高成年小鼠的平均体重[3]。Chow EK等结合SB转座子系统和水动力法尾静脉注射(Hydrodynamic tail vein injection, HTVI)在小鼠肝脏中通过过表达原癌基因Myc诱导肝癌形成[4]。
通过基于CRISPR/Cas9的高通量负向筛选技术和自主构建的靶向泛素蛋白酶体系统相关基因的sgRNA亚文库对人肝癌HepG2细胞株进行高通量负向筛选(见图1),发现过氧化物酶体生物合成因子2(Peroxisomal Biogenesis Factor 2,PEX2)敲除后可以抑制HepG2细胞生长。PEX2是过氧化物酶体的一种膜蛋白,与PEX10、PEX12构成E3泛素连接酶复合体,通过泛素化PMP70和PEX5参与过氧化物酶体自噬活动[5]。进一步研究发现PEX2基因的缺失可导致肝癌细胞中过氧化物酶体功能丧失、多种代谢途径紊乱,同时可抑制mTOR(The mammalian target of rapamycin)信号转导通络,最终引起肝癌(Liver cancer)细胞发生自噬和死亡[6]。
为进一步了解PEX2在体内对肝癌发生发展的作用,我们运用睡美人( Sleeping Beauty, SB) 转座系统构建小鼠肝癌模型,即通过水动力法尾静脉注射向小鼠肝脏导入携带原癌基因MYC的转座子质粒pT3EF1α-c-Myc和表达转座酶SB100X的pCMV(CAT)T7-SB100 质粒,其中转座酶通过识别转座子中MYC表达结构两侧的反向重复序列,将其剪切并整合到小鼠肝脏细胞的基因组中,可以实现MYC在整合细胞中的稳定表达。运用该种方法成功地在小鼠体内诱导出肝癌。随后,我们构建了同时含有针对PEX2的shRNA序列或对照shNC序列和原癌基因MYC的重组转座子系统,以探索PEX2在体内对肝癌形成的影响。

图1 基因筛选模式图Figure 1 Genetic screening model
1 材料与方法
1.1 材料
1.1.1 质粒、菌株、细胞和小鼠 质粒pT3EF1α-c-Myc,pCMV(CAT)T7-SB100 购自Addgene公司。质粒pLKO.1-shNC和pLKO.1-shPEX2由北京国家蛋白质研究中心分子遗传实验室保存。大肠杆菌Mach1-T1感受态细胞购自博迈德公司。小鼠肝癌细胞株Hepa1-6由北京国家蛋白质研究中心分子遗传实验室保存。6-8周龄小鼠FVB购买于北京维通利华实验动物技术有限公司。
1.1.2 工具酶和试剂 各种内切酶和 T4 连接酶均购自 NEB公司, 质粒提取试剂盒和凝胶回收试剂盒为OMEGA品牌,Lipofectamine 2000 购自Thermo Fisher Scientific 公司。
1.2 方法
1.2.1 载体pT3EF1α-GFP的构建 以载体pENTRY-EF1α-GFP-Mir为模板、F-EF1α-GFP-AgeI和R-EF1α-GFP-Not I为引物PCR扩增EF1α-GFP序列(见表1),琼脂糖凝胶电泳鉴定并回收PCR产物。PCR 反应体系为:1μL pENTRY-EF1α-GFP-Mir、1.5μL引物、25μL 2×PCR Buffer for KOD FX、10μL 2mM dNTPs、1μL KOD FX、10μL ddH2O。反应条件为:94℃预变性 2min;先 98℃变性 10s、 64℃退火 20s、 68℃延伸 90s,重复变性、退火、延伸30个循环;最后68℃延伸 10min 终止反应。
用AgeI 和Not I同时酶切载体pT3EF1α-c-Myc和EF1α-GFP片段,琼脂糖凝胶电泳回收酶切产物,并对产物进行T4连接,获得目标载体pT3EF1α-GFP。酶切体系为:40μL pT3EF1α-c-Myc或EF1α-GFP片段、5μL cutsmart、2μL AgeI、2μL Not I、1μL ddH2O。T4连接体系:0.5μL 酶切后pT3EF1α-c-Myc、1.5μL 酶切后EF1α-GFP片段、0.5μL T4连接酶、0.5μL T4连接缓冲液、2μL ddH2O。对获得的载体进行转化、挑取单克隆、提质粒并进行测序,以鉴定载体的正确性。质粒提取严格按照OMEGA小提试剂盒步骤进行操作。质粒测序委托北京擎科新业生物有限公司进行。
1.2.2 质粒表达检测 小鼠肝癌细胞Hepa1-6接种于6孔培养板中,待密度为40%~60%时,用Lipofectamine 2000进行质粒转染,pT3EF1α-c-Myc和pT3EF1α-GFP质粒各转染2μg,并于48小时后收取样品,提取RNA,通过qPCR实验测定载体pT3EF1α-c-Myc和pT3EF1α-GFP中MYC的表达。
1.2.3 SB转座子介导的MYC过表达诱导小鼠肝癌预实验 实验组与对照组各五只FVB小鼠,对照组注射转座子质粒 pT3EF1α-GFP和转座酶SB100X表达载体pCMV(CAT)T7-SB100 ;实验组注射转座子质粒 pT3EF1α-c-Myc和转座酶SB100X表达载体pCMV(CAT)T7-SB100 。质粒使用量为:20μg/只,其中转座子与转座酶载体比例为25∶1。质粒溶于格林溶液。注射方法采用水动力法尾静脉注射(Hydrodynamic tail vein injection, HTVI),即在5~9s内将质粒溶液通过小鼠尾静脉注射入小鼠体内。
1.2.4 载体pT3EF1α-c-Myc-shNC、pT3EF1α-c-Myc-shPEX2的构建 设计合成小鼠PEX2基因的shRNA序列shPEX2以及对照组shNC(见表2),并将其连接入载体pLKO.1中构建pLKO.1-shPEX2、pLKO.1-shNC载体。在小鼠肝癌细胞Hepa1-6中验证其敲低PEX2的作用。以载体pLKO.1-shNC和pLKO.1-shPEX2为模板,F-U6-shNC-NsiI、R-U6-shNC-PacI和 F-U6-shPEX2-NsiI、R-U6-shPEX2-PacI为引物扩增U6-shNC和U6-shPEX2片段(见表1),琼脂糖凝胶电泳鉴定并回收PCR产物。PCR 反应体系为: 1μL pLKO.1-shNC载体或pLKO.1-shPEX2、1.5μL引物、 25μL 2×PCR Buffer for KOD FX、10μL 2mM dNTPs、1μL KOD FX、10μL ddH2O。反应条件为:94℃预变性 2min;先 98℃变性 10s、 64℃退火 20s、 68℃延伸 20s,重复变性、退火、延伸30个循环;最后68℃延伸 10min 终止反应。
用NsiI 和PacI同时酶切载体pT3EF1α-c-Myc和U6-shNC、U6-shPEX2片段,琼脂糖凝胶电泳回收酶切产物,并对产物进行T4连接,获得目标载体pT3EF1α-c-Myc-shNC和pT3EF1α-c-Myc-shPEX2。酶切体系:40μL pT3EF1α-c-Myc或U6-shNC片段或U6-shPEX2片段、5μL cutsmart、2μL NsiI、2μL PacI、1μL ddH2O。T4连接体系:0.5μL 酶切后pT3EF1α-c-Myc、1.5μL 酶切后U6-shNC片段或U6-shPEX2片段、0.5μL T4连接酶、 0.5μL T4连接缓冲液、2μL ddH2O。
对上述获得的载体进行转化、挑取单克隆、提质粒并进行测序,以鉴定载体的正确性。质粒提取严格按照OMEGA小提试剂盒步骤进行操作。质粒测序委托北京擎科新业生物有限公司进行。
1.2.5 pT3EF1α-c-Myc-shNC、pT3EF1α-c-Myc-shPEX2质粒表达检测 小鼠肝癌细胞Hepa1-6接种于 6 孔培养板中,待密度为40%-60%时,用Lipofectamine 2000进行质粒转染,每种质粒各转染2μg,并于48小时后收取样品,提取RNA,通过qPCR实验测定实验组载体pT3EF1α-c-Myc-shPEX2的表达是否实现PEX2基因的敲低。

表1 载体构建引物Table 1 The primers for vector

表2 shRNA序列Table 2 The sequence of shRNA
2 结果
2.1 载体pT3EF1α-GFP的构建 以载体pENTRY-EF1α-GFP-Mir为模板扩增EF1α-GFP片段,经琼脂糖凝胶电泳检测,电泳条带清晰可见,并且与预期一致,大小为1919bp(见图2A)。用AgeI 和NotI同时酶切载体pT3EF1α-c-Myc和EF1α-GFP片段,然后进行琼脂糖凝胶回收,并用T4连接酶对产物进行连接,通过转化对载体进行筛选,挑取单克隆并提取质粒进行酶切和测序以鉴定载体的正确性。酶切鉴定显示pT3EF1α-GFP经AgeI 和NotI酶切后,获得大小为3935bp和1912bp的条带(见图2B),并且测序结果显示GFP与原序列吻合,说明克隆所得的载体 pT3EF1α-GFP正确。
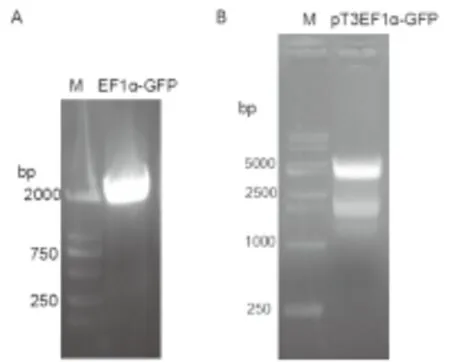
图2 PCR产物(A)与质粒pT3EF1α-GFP酶切鉴定(B)
Figure2TheproductsofPCR(A)andDetectionofpT3EF1α-GFPvector(B)
2.2 pT3EF1α-GFP和pT3EF1α-c-Myc载体中MYC基因的表达情况 用Lipofectamine 2000转染法分别将2μg转座子质粒pT3EF1α-GFP和pT3EF1α-c-Myc转染入小鼠肝癌细胞Hepa 1-6中,并于48小时后收取样品提取RNA检测RNA水平MYC基因的表达情况。结果显示,载体pT3EF1α-c-Myc中存在MYC表达,而载体pT3EF1α-GFP中无MYC表达。表明可用pT3EF1α-GFP转座子作为对照,以比较显示原癌基因MYC诱导肝癌形成的作用,见图3。
2.3 SB转座子介导的MYC过表达诱导小鼠肝癌预实验 借助SB转座子系统在肝脏细胞中通过过表达原癌基因MYC以诱导肝癌形成。向实验组注射携带MYC基因的pT3EF1α-c-Myc转座子质粒和表达转座酶SB100X的pCMV(CAT)T7-SB100 质粒,转座酶通过识别转座子载体中MYC表达结构两侧的反向重复序列(ITRs),将其剪切并重新整合到小鼠肝脏细胞的基因组中,可以实现MYC在整合细胞中的稳定表达。对照组注射携带对照基因GFP的pT3EF1α-GFP转座子质粒和表达转座酶SB100X的pCMV(CAT)T7-SB100 质粒,以相同的机制,最终被整合入小鼠肝脏细胞基因组中的为GFP基因。实验结果显示,从注射后第5周开始,实验组小鼠出现荷瘤死亡,而对照组小鼠未见成瘤(见图4)。我们成功诱导出肝癌,证明SB转座系统介导的MYC过表达诱导肝癌形成技术可行。
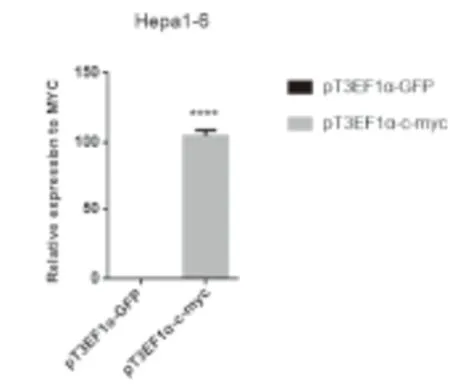
图3 质粒pT3EF1α-GFP与pT3EF1α-c-Myc中MYC的表达
Figure3ExpressionofMYCinpT3EF1α-GFPandpT3EF1α-c-Myc

图4 SB转座系统介导MYC诱导小鼠肝癌形成
Figure4SBtransposonmediatedMYC-inducedlivercancerformationinmice
注:A.小鼠正常肝脏组织;B.小鼠肝癌组织
2.4 载体pT3EF1α-c-Myc-shNC、pT3EF1α-c-Myc-shPEX2的构建 集落形成实验显示以pLKO.1-shNC为对照,载体pLKO.1-shPEX2在小鼠细胞Hepa1-6中敲低PEX2有效,可抑制该细胞的增殖(见图5),证明该shPEX2序列可用。然后以载体pLKO.1-shNC和pLKO.1-shPEX2为模板扩增U6-shNC和U6-shPEX2片段,经琼脂糖凝胶电泳检测,电泳条带清晰可见,并且与预期一致,U6-shNC大小为318bp,U6-shPEX2大小为322bp(见图6A)。用NsiI 和PacI同时酶切载体pT3EF1α-c-Myc和U6-shNC、U6-shPEX2片段,然后进行琼脂糖凝胶回收,并用T4连接酶对产物进行连接,通过转化对载体进行筛选,挑取单克隆并提取质粒进行酶切和测序以鉴定载体的正确性。酶切鉴定显示载体pT3EF1α-c-Myc-shNC和pT3EF1α-c-Myc-shPEX2经NsiI 和PacI酶切后,pT3EF1α-c-Myc-shNC获得大小为6472 bp和310 bp两个条带,pT3EF1α-c-Myc-shPEX2获得大小为6472 bp和314 bp两个条带(见图6B),并且测序结果显示U6-shNC和U6-shPEX2与原序列吻合,说明克隆所得的载体pT3EF1α-c-Myc-shNC、pT3EF1α-c-Myc-shPEX2正确,载体图谱,见图7。
2.5 载体中PEX2基因的敲低情况 用Lipofectamine 2000转染法分别将2μg转座子质粒转染小鼠肝癌细胞Hepa 1-6,并于48小时后收取样品提取RNA检测RNA水平上PEX2基因的敲低。结果显示,载体pT3EF1α-c-Myc-shPEX2相较于对照载pT3EF1α-c-Myc-shNC,PEX2基因敲低约40%,见图8。
3 讨论
肝癌是目前全球癌症相关死亡的第二大常见原因[7],成功构建肝癌动物模型,对探索影响肝癌形成的分子功能进而研究人类肝癌的发生发展具有重要现实意义。小鼠因遗传物质跟人类相似,有繁殖能力强、生命周期短、基因修饰方便等特点,成为重要的模式生物。通过多种方法构建小鼠肝癌模型,可为研究肝癌相关的药物筛选、肝癌发生机制等提供实验载体。利用化学物质可诱发肝癌形成,匡志鹏等通过联合采用二甲基亚硝胺(DEN)/四氯化碳(CCl4)/乙醇成功诱导C57BL/6J小鼠肝癌形成[8]。Vartanian V等运用黄曲霉毒素(AFB1)诱导出小鼠肝癌[9]。化学物质诱发肝癌虽在研究中多见,但其致死率较高,并且诱发时间较长。借助人类或者小鼠肝癌组织或者肝癌细胞可构建小鼠移植肝癌模型。移植部位常见于背侧皮下,肝脏等。张建民等采用直接注射法,将鼠肝癌H22细胞注射入小鼠肝脏,术后两周观察发现实验小鼠均可见肿瘤生长[10]。采用移植法构建小鼠肝癌模型成瘤率高,实验周期短且个体差异小,但也存在问题。皮下移植瘤因缺乏肝脏特异的生长环境,导致皮下移植肿瘤的生物学行为与肝脏原位肿瘤存在差异,同时移植瘤多构建在免疫力缺陷的裸鼠身上,因此此种模型不适用于肝癌发展与免疫之间关系的研究。随着转基因技术的发展,借助转基因技术构建小鼠肝癌模型也应运而生。人类肝癌的80%是由乙型肝炎病毒(HBV)和或丙型肝炎病毒感染(HCV)所致,借助转基因技术,已成功构建出携带肝炎病毒编码基因的小鼠模型。Chisari FV等通过向单个小鼠受精卵中显微注射含有HBV表面抗原(HBsAg)和前S和X抗原编码区的HBV DNA亚基因组片段,成功构建HBV基因携带的小鼠模型[11]。Kyoji Moriya等构建的HCV核壳蛋白转基因小鼠,在16个月的时候发展出肝细胞癌[12]。HBV、HCV慢性携带小鼠模型的构建,有助于肝炎病毒相关的肝脏疾病的研究。同时,结合转基因技术将原癌基因导入小鼠肝脏组织,通过原癌基因的表达作用诱导正常肝脏细胞癌变,最终形成肝癌的方法也已成功实现。B Xin等借助水动力尾静脉注射(HTVi)技术和SB转座子系统,将AKT和HRAS基因导入C57BL/6J小鼠肝脏细胞中,并于大约8周时诱导出肝癌[13],并且发现MYC基因的活化在AKT和HRAS基因诱导肝癌形成中起着重要作用。Pin Liu等借助水动力尾静脉注射(HTVi)技术和SB转座子系统,将原癌基因myc导入FVB/N 小鼠肝脏细胞中,注射后约6周小鼠出现荷瘤死亡,同时发现mTORC1信号通路活化在MYC诱导小鼠肝癌形成中的关键作用[14]。通过水动力尾静脉注射(HTVi)技术将质粒注射入小鼠体内,其中肝脏为质粒的主要获取器官,心脏、肺、肾、脾等的质粒获取不足肝脏获取的0.1%,约10%~40%肝脏细胞可获取质粒[15],因此水动力尾静脉注射可达到很好的肝脏靶向性。利用SB转座子技术可实现目标基因在宿主基因组中的整合,以实现目的基因是长期稳定表达。结合这两种技术可将目的基因整合入肝脏细胞,但最终获取并能稳定表达目的基因的肝脏细胞只有2%~10%[15]。肿瘤细胞周围围绕正常肝脏细胞,更加符合人类肝癌的发展模式。借助水动力尾静脉注射(HTVi)技术和SB转座子系统使肝脏细胞过表达原癌基因MYC,从而诱发肝癌形成的方法现已比较成熟,并且操作方便、实验周期短。需要注意的是,遵照SB转座子系统的机制,构建重组转座子时,为实现目标序列能够被整合到受体基因组中,重组序列必须插入在两侧反向重复序列之间。
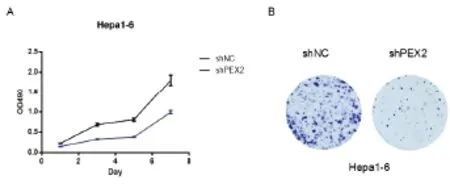
图5 shPEX2的表达可抑制Hepa1-6细胞的增殖Figure 5 The expression of shPEX2 can inhibit the proliferation of Hepa1-6 cells

图6 PCR产物(A);pT3EF1α-c-Myc-shNC、pT3EF1α-c-Myc-shPEX2酶切鉴定(B)Figure 6 The products of PCR (A) ; Detection of pT3EF1α-c-Myc-shNC vector and pT3EF1α-c-Myc-shPEX2 vector (B)
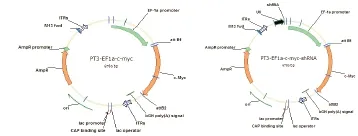
图7 质粒pT3EF1α-c-Myc和pT3EF1α-c-Myc-shRNA图谱Figure 7 Map of pT3EF1α-c-Myc and pT3EF1α-c-Myc-shRNA

图8质粒pT3EF1α-c-Myc-shPEX2可减少Hepa1-6细胞中PEX2的表达
Figure8PlasmidpT3EF1α-c-Myc-shPEX2ReducesPEX2ExpressioninHepa1-6Cells
4 结论
本研究借助 HTVI技术和SB转座系统过表达c-Myc成功诱导小鼠肝癌形成,并在SB转座子质粒pT3EF1α-c-Myc基础上成功构建了可在小鼠肝癌细胞Hepa1-6中敲低PEX2基因的重组转基因载体,以探索PEX2在体内对肝癌发生发展的影响。
猜你喜欢
华人时刊(2022年9期)2022-09-06
林业科学(2022年1期)2022-03-23
浙江农林大学学报(2021年3期)2021-07-12
江西农业学报(2021年4期)2021-04-20
水生生物学报(2021年1期)2021-02-04
华人时刊(2020年15期)2020-12-14
三农资讯半月报(2020年11期)2020-06-21
中国农业科学(2020年7期)2020-04-11
浙江农林大学学报(2016年6期)2016-12-12
中国当代医药(2015年9期)2015-03-01
