
稻米中赤霉酸的HPLC测定方法研究
2018-09-18 02:57胡银川李明元刘秋凤徐珑珀
西华大学学报(自然科学版) 2018年5期
胡银川,李明元,刘秋凤,王 涛,向 昕,徐珑珀,钟 洋,谭 欣
(1.四川省食品药品检验检测院,四川 成都 610097; 2.西华大学食品与生物工程学院,四川 成都 610039)
赤霉素是一类促进植物和果实生长的内源激素,其中以赤霉酸(GA3)的应用最为广泛[1-2]。赤霉酸能促进种子发芽、植物和果实生长,被广泛应用于农作物的栽培[3-4]。水稻喷施赤霉酸可促进水稻生长,提高抽穗率和成穗率,使稻谷颗粒饱满,提高水稻产量[5]。然而,长时间摄入赤霉酸会影响人体生长发育,危害人体健康[6]。欧美规定赤霉酸在农产品中的最高残留限量值为0.2 mg/kg,而我国尚未出台相应的国家标准;因此,建立起高效、准确的赤霉酸检测方法对我国农产品出口以及人民健康具有极其重要的意义[7]。由于稻米基体复杂,且赤霉酸在稻米中的含量极低;因此,需要选择一种精确、可靠的分析方法[8-9]来测定稻米中赤霉酸含量。赤霉酸的检测方法主要有分光光度法、薄层色谱法、毛细管电泳法和气相色谱法等[10-11]。由于分光光度法、薄层色谱法、毛细管电泳法达不到对赤霉酸的痕量测定的要求,且气相色谱法前处理过程中的衍生较繁琐;因此,本文采用高效液相色谱仪对稻米中赤霉酸进行检测[12]。
1 材料与方法
1.1 仪器与试剂
LC-20A高效液相色谱仪,日本岛津公司;TB-214电子天平,北京赛多利斯仪器系统有限公司;TDL-40B离心机,上海安亭科学仪器厂;Milli-Q超纯水仪,Millipore;KQ-100DE数控超声波清洗器,昆山市超声仪器有限公司;SFG-02.400电热恒温鼓风干燥箱,黄石市恒丰医疗器械有限公司;ZHWY-200D恒温振荡器,上海智城分析仪器制造有限公司;ZN-200A粉碎机,长沙市岳麓区中南制药机械厂;RE-52B转蒸发仪,上海亚荣生化仪器厂;pHS-3C酸度计,成都世纪方舟科技有限公司;GC-17真空泵,浙江黄岩求精真空泵厂;XW-80A型漩涡混合器,常州诺基仪器有限公司;Discovery C18色谱柱(250 mm×4.6 mm×5 μm),美国Supelco公司。
赤霉酸标准品(纯度大于99.9%),德国Dr.Ehrenstorfer公司;乙酸乙酯、甲醇、石油醚、异丙醇均为色谱纯,Fisher Scientific公司;盐酸为优级纯,成都科龙化工试剂厂;试验用水为超纯水。
1.2 样品前处理
将稻米粉碎,称取10 g稻米置于锥形瓶中,加入80%甲醇溶液50 mL,振荡提取18 h,过滤样品。向滤渣中加入80%甲醇溶液30 mL,超声提取30 min,过滤样品,合并2次滤液。移取20 mL滤液,分别用10 mL石油醚脱色2次,旋转蒸发至5 mL,加入5 mL乙酸乙酯进行萃取,漩涡混匀30 s,取上层液过无水硫酸钠小柱,再用乙酸乙酯重复萃取3次,氮吹至近干,用2 mL甲醇定容,用0.45 μm有机滤头过滤,上机分析。
1.3 色谱条件
流动相:V甲醇∶V水(30 ∶70);流动相pH值为4.0;流速为1.0 mL/min;柱温为40 ℃;检测波长为210 nm;进样量为10 μL。
2 结果与讨论
2.1 检测波长的选择
对赤霉酸标准溶液进行全波段扫描,扫描光谱图见图1。由图1可知,赤霉酸在200、210、225和655 nm均有吸收峰。 225和655 nm吸收强度较低,200 nm时流动相甲醇吸收强度会明显增强,对检测造成干扰,故选用210 nm作为检测波长。
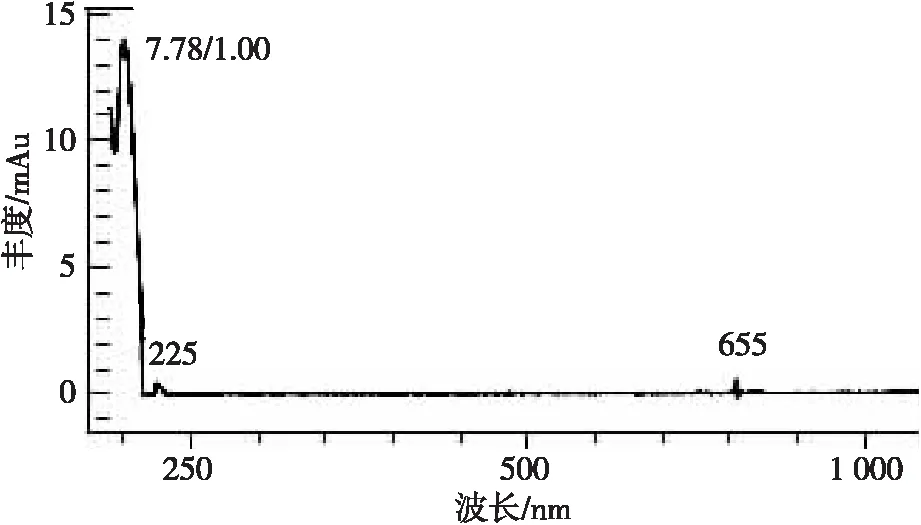
图1 赤霉酸扫描光谱图
2.2 流动相的选择
在柱温为35 ℃,流速为1.0 mL/min,流动相pH值为4.0,进样量10 μL条件下,考察流动相(V甲醇∶V水为30 ∶70、V甲醇∶V水为35 ∶65、V甲醇∶V水为40 ∶60)对分析的影响。当V甲醇∶V水为30 ∶70时,峰形尖锐且峰宽小,故流动相选择V甲醇∶V水为30 ∶70。
2.3 流动相流速的选择
在柱温35 ℃,流动相V甲醇∶V水为30 ∶70,流动相pH值为4.0,进样量10 μL条件下,考察流动相流速(0.6、0.8、1.0 mL/min)对分析的影响。流动相在1.0 mL/min时,峰形尖锐,保留时间短,故流动相流速选择1.0 mL/min。
2.4 柱温的选择
在流动相V甲醇∶V水为30 ∶70,流速为1.0 mL/min,流动相pH值为4.0,进样量10 μL条件下,考察柱温(30、35、40 ℃)对分析的影响。当柱温为40 ℃时,赤霉酸的色谱峰分离度较好,出峰时间最短,缩短了分析时间,故柱温选择40 ℃。
2.5 流动相pH值的选择
在流动相V甲醇∶V水为30 ∶70,流速为1.0 mL/min,进样量10 μL条件下,考察流动相pH值(3.0、4.0、5.0)对分析的影响。当pH值为4.0时,峰形尖锐,分离度较好,故流动相pH值选择4.0。
2.6 线性关系与检出限
取赤霉酸系列标准溶液(5、10、20、40、80、160、320 μg/mL),按照上述色谱条件进行分析,外标法定量。在5 ~320 μg/mL的线性范围内,具有良好的线性关系(R2=0.999 96)。最低检出限为2.0 μg/g (S/N=3)。图2为200 μg/mL的赤霉酸标准品的色谱图。图3为稻米中阳性样品色谱图。
2.7 精密度
称取12份稻米样品,进行样品前处理,每次进样10 μL,测定稻米中赤霉酸含量,结果见表1。由表1可知,12次测定值的相对标准偏差(RSD)为3.2%,表明本方法具有良好的精密度。

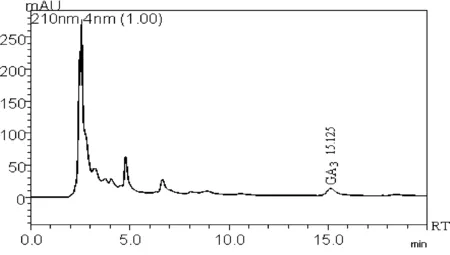
图2 赤霉酸标准品色谱图 图3 稻米中阳性样品色谱图

表1 方法精密度(n=12)
2.8 回收率
本文采用加标回收实验评价本方法的可靠性,对稻米样品进行了低、中、高3个不同水平添加回收实验,每一水平分别做6次平行实验,计算平均回收率。称取本底含量为6.46 μg/g的稻米样品,进行添加质量分数为5.0、15.0和30.0 μg/g的加标回收实验,每个水平做6次平行实验,算出其平均回收率,结果见表2。由表2可知,赤霉酸的回收率在91.7%~98.7%之间,相对标准偏差在1.5%~2.9%之间,说明本方法准确可靠。

表2 加标回收实验结果(n=6)
3 结论
本文建立的稻米中赤霉酸的HPLC测定方法为:称取10.0 g粉碎的稻米置于锥形瓶中,加入80%甲醇溶液50 mL,振摇18 h,过滤样品,向滤渣中加入80%甲醇溶液30 mL,超声提取30 min,过滤样品,合并2次滤液,移取20 mL滤液,分别用10 mL石油醚脱色2次,旋转蒸发至5 mL,加入5 mL乙酸乙酯进行萃取,漩涡混匀30 s,取上层液过无水硫酸钠小柱,再用乙酸乙酯重复萃取3次,旋转蒸发至干,用2 mL甲醇复溶,用0.45 μm有机滤头过滤,HPLC分析。结果表明:在5~320 μg/mL范围内具有良好的线性关系R2=0.999 96,最低检出限为2.0 μg/g;精密度好,在12次平行实验测定值的相对标准偏差为3.2%;在5.0、15.0和30.0 μg/g添加水平下,赤霉酸的回收率在91.7%~98.7%之间。
猜你喜欢
初中生学习指导·提升版(2022年4期)2022-05-11
美食(2022年5期)2022-05-07
中学生数理化·八年级物理人教版(2022年4期)2022-04-26
早期教育(家庭教育)(2021年11期)2021-12-17
少儿科学周刊·儿童版(2021年21期)2021-12-11
粮食储藏(2021年6期)2021-03-29
大众科学(2020年7期)2020-10-26
中国粮食经济(2018年5期)2018-12-27
小天使·六年级语数英综合(2018年1期)2018-10-08
——CIPAC分析手册 O卷的分析方法
农药科学与管理(2018年2期)2018-08-07
