
基于RNA-seq的基因训练集构建方法
2018-09-10 10:14段荣静刘金定
数据采集与处理 2018年4期
段荣静 刘金定
(1. 南京农业大学领域知识关联研究中心,南京,210095; 2. 南京农业大学生物信息学中心,南京,210095)
引 言
从头基因预测软件(下简称预测软件)常用于识别基因组上的编码基因结构,在基因组注释中发挥着非常重要的作用。预测软件使用前必须优化其模型参数使之适应被测序物种。除了几个非监督预测软件外[1],大多数预测软件都属于监督训练软件[2-5],其训练过程必须建立在足够数量的可靠基因结构上。对于新测序的基因组而言,其研究基础薄弱,往往缺乏足够数量的编码基因结构用于训练,这大大限制了预测软件的使用。因此,构建高质量的基因训练集是一个非常有价值的工作。
为新测序物种构建基因训练集(下简称训练集)是一个非常耗时、费力的过程,有时甚至比基因组测序本身还繁琐。构建训练集不仅需要克隆全长基因,而且还需要专家人工矫正基因结构。另外,由于高表达和高保守的基因容易被发现和验证,所以人工克隆方法构建的训练集通常会出现样本不平衡的现象。考虑到人工准备基因训练集的繁琐和缺陷,很多基因组注释项目直接用其他近缘物种基因对预测软件进行训练,或者直接使用其他物种模型参数跳过预测软件训练环节。预测软件性能依赖物种特异性的模型参数,而物种间的差异性又是必然存在的,因此这种做法必然会导致预测性能下降,尤其在亲缘关系较远物种之间更是如此。为此,研究人员提出在基因组上识别保守基因结构用于构建训练集的方法[6-8]。这些方法定义了真核生物共同拥有的核心蛋白家族并提供了识别这些蛋白对应基因结构的计算方法。这些方法存在两个缺陷:(1)真核生物核心基因过于保守,导致训练集不能特异优化被测序物种的模型参数;(2)测序基因组不完整导致基因组上核心基因丢失,从而引发训练基因数量不足的现象。RNA-seq是快速获得基因表达数据的有效方法,已成为基因组注释项目中必不可少的步骤。理论上,RNA-seq可以获得全部表达的基因序列,而且通过组装读段可以获得全长序列[9-11]。将测序读段比对到基因组上,可以获得大量外显子内含子位置信息,进一步组装将得到外显子-内含子相互串联的基因结构。但是由于测序和组装错误的存在,导致获得的基因结构质量不能满足构建训练集的要求[12]。另一方面,基于RNA-seq组装的基因结构无法直接给出编码区的结构以及起始密码子和终止密码子位置信息,因此进一步降低了组装结果的可用性。
针对基因组新测序物种基因匮乏,难以满足预测软件训练需要的现状,本文提出了一种以新测序物种自身RNA-seq组装为基础,结合蛋白同源证据的可靠基因训练集构建方法(Building reliable training gene set,BRTGS)。该方法利用蛋白同源证据排除不完整和错误的RNA-seq组装结构,利用RNA-seq组装结构弥补蛋白同源证据难以获得同源区外基因结构以及起始密码子和终止密码子位置的缺陷。将该方法应用于果蝇、人、拟南芥和水稻等几种模式生物基因组上,实验结果表明该方法不仅可以为各种组装水平的基因组构建足够数量的高质量训练基因,而且训练后的预测软件可以获得很好的预测性能。
1 从头基因预测软件训练以及相关数据基础
1.1 从头基因预测软件训练
从头预测软件训练是指利用物种已有的基因结构信息优化其模型参数,使之适应该物种。虽然预测软件训练效果会随着训练基因的数量增加而逐步提高,但实践表明当训练基因数量超过1 000 h时,训练效果的提升不再明显。因此,构建基因训练集时更注重基因质量而不是数量。高质量的训练基因不仅要具有完整的外显子-内含子串联结构,而且要有正确的起始密码子和终止密码子位置信息(图1)。RNA-seq组装可以快速获得大量基因外显子-内含子串联结构信息,但是这些结构信息不仅存在组装错误,而且缺少起始密码子和终止密码子位置信息,因此不能直接用于构建基因训练集。
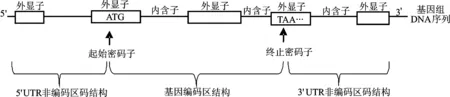
图1 真核基因结构Fig.1 Structure of eukaryotic genes
1.2 RNA-seq组装中的编码基因结构
目前公共数据库中存在大量低N50基因组,甚至存在N50为10K的基因组。从头预测软件训练一般需200~1 000个可靠的基因结构。为了模拟各种组装水平的基因组,果蝇、人、拟南芥和水稻等几种模式生物基因组被打碎随机抽取序列构成N50分别为2K,5K,10K,20K,40K,80K,160K,320K,640K和1M的片段化基因组(这些基因组在下文被分别引用为G2K,G5K,G10K,G20K,G40K,G80K,G160K,G320K,G640K和G1M)。在低组装水平的基因组中大量基因组序列没有被组装到一起,导致跨度较大的基因被分散在多个基因组序列中而被重复计数,因此随着N50下降,基因总数不断提高(图2(a)),但完整基因数量不断下降(图2(b))。在4个物种中,人类基因平均跨度最大,所以基因被碎片化的程度最高。在G2K基因组中,人类基因总数上升比例最高达到了460%,而完整基因数量下降最多达到42%。从数字上看,在G2K基因组中,4个物种分别保留了8 228,10 003,16 349和22 431个完整基因,这表明组装水平较低的基因组中仍然保留着大量完整基因资源可用于构建基因训练集。
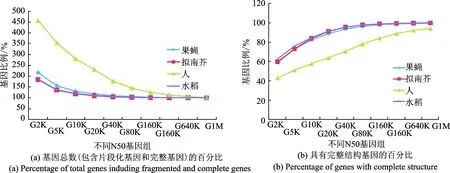
图2 N50对保留在片段化基因组中基因数量的影响Fig.2 Influence of genome N50 on the numbers of genes retained in fragmented genomes
虽然低水平组装的基因组中仍然存在大量完整基因结构,但需要确定RNA-seq组装结果是否能够提供足够数量的正确外显子-内含子串联结构(简称基因结构)。鉴于生物学实验通常3个重复的要求,本文随机地为每个物种准备了3个RNA-seq数据。首先将每个RNA-seq数据比对到基因组上,然后组装比对结果获得基因结构。一般而言,将多个RNA-seq组装结果合并将得到更多基因结构(基因结构数量并不总是随着RNA-seq数量增加而增长,当RNA-seq测序总量足够饱和时,基因结构数将不再增长),因此,本文进一步利用CuffMerge将3个RNA-seq组装结果合并在一起产生1个非冗余组装结果,这样每个基因组上得到4个RNA-seq组装结果。由于绝大多数预测软件只预测编码区部分,所以本文只统计编码区部分的结构被完整且正确组装的数量结果如表1所示。第1个样品RNA-seq数据在G2K基因组上组装获得正确编码基因结构数量最少,组合3个RNA-seq数据在G1M上组装获得正确编码基因结构数量最多。由于这两种情况分别代表了最差组装结果和最优组装结果,因此本文后续研究只关注这两种RNA-seq组装结果。在G2K基因组上,第1个样品RNA-seq组装结果分别为4个物种提供了2 482,6 627,5 649 和7 928个正确编码基因结构。将3个样品RNA-seq组装结果合并起来,为G2K基因组分别提供4 933, 10 941, 8 764和11 846个正确编码基因结构。对于G1M基因组而言,无论是第1个样品RNA-seq还是组合RNA-seq都组装出非常多的正确编码基因结构。果蝇的第1个样品RNA-seq数据在G1M上组装的正确编码基因结构数量最少为3 866个,人类的组合RNA-seq数据在G1M上组装的正确编码基因结构数量最多达到19 535个。预测软件的训练只需几百个正确编码基因结构,从RNA-sq组装结果上看,即使在低组装水平的基因组(G2K)上采用单个RNA-seq数据,也能组装出近2 500个正确编码基因结构,这表明基于RNA-seq的组装结果可为预测软件训练提供充足的编码基因结构。

表1 RNA-seq组装的完整编码基因结构的数量
注:每个物种的样品1、样品2和样品3对应的具体RNA-seq见3.1节实验数据。
2 基于RNA-seq组装的基因训练集构建方法
2.1 包含完整编码区的RNA-seq转录本识别
首先用Tophat将RNA-seq短读段序列比对到基因组上,然后用Cufflinks对比对结果进行组装获得基因结构,最后利用CuffMerge对每个基因组的3个RNA-seq组装结果进行合并获得非冗余基因结构。根据RNA-seq组装的基因结构获得对应的转录本序列后,用局部比对搜索工具X(Basic local alignment search tool X, BLASTX)将转录本序列比对到参考蛋白数据库中(E-value设为1e-10),只保留前50个洞(Gap)百分比小于3%且对齐区域覆盖参考蛋白序列75%以上的比对结果用于后续分析。如果一个基因结构符合上述比对结果,那么该基因结构将被认为可能包含完整编码区。
2.2 起始密码子和终止密码子位置范围计算
正确识别起始密码子和终止密码子位置对提升训练集质量具有重要的意义,为此BRTGS进一步利用同源参考蛋白序列初步确定起始密码子和终止密码子位置范围,以提高后续起始密码子和终止密码子的预测可靠性,结果如图3所示。转录本上的每个比对结果都可以推导一对起始密码子和终止密码子位置。在转录本对齐起始位置向5端延伸一定长度可以推导出起始密码子位置,这个长度为参考蛋白序列上N端非对齐氨基酸长度的3倍。同样,在转录本对齐终止位置向3端延伸一定长度可以推导出终止密码子位置。一个转录本所有比对结果推导出的起始密码子和终止密码子位置收集在一起就构成起始密码子和终止密码子位置范围。

图3 计算起始密码子和终止密码子范围Fig.3 Calculation of site range of start codon and stop codon
2.3 转录本起始密码子和终止密码子位置确定
根据起始密码子和终止密码子确定方法,可将包含完整编码结构的转录本(Complete coding transcripts, CCTs)分为以下4类:
(1)CCT I :如果最佳比对结果的对齐区域完全覆盖参考蛋白序列,对齐区域5端的3个碱基为起始密码子,3端向后3个碱基为终止密码子,那么这个转录本属于CCT I,转录本上的对齐区域为编码区。
(2)CCT II:如果最佳比对结果的对齐区域覆盖参考蛋白序列的N端,对齐区域5端的3个碱基为起始密码子,3端向后出现的第1个终止密码子位于终止密码子范围内,那么这个转录本属于CCT II。转录本的编码区从对齐区域开始到推导的终止密码子上游位置结束。
(3)CCT III:如果最佳比对结果的对齐区域覆盖参考蛋白序列的C端,3端向后3个碱基正好是终止密码子,用ATGpr[13]计算推导出的起始密码子位于起始密码子位置范围内,那么这个转录本属于CCT III。转录本的编码区为从ATGpr计算的起始密码子位置开始到终止密码子上游位置结束。
(4)CCT IV:如果最佳比对结果的对齐区域既不能覆盖参考蛋白序列的N端,也不能覆盖C端,但是用ATGpr计算推导出的起始密码子位于起始密码子位置范围内,下游第1个终止密码子位于终止密码子范围内,那么这个转录本属于CCT IV。转录本的编码区为从ATGpr计算的密码子位置开始到推导的终止密码子上游位置结束。
在确定起始密码子和终止密码位置后,再次计算转录本编码区的长度,如果编码区长度大于最佳比对的参考蛋白序列长度130%,那么这个转录本将被淘汰。
2.4 同源聚类后排序生成基因训练集
在构建的4类CCTs中,CCT I的起始密码子和终止密码同时具有表达、同源和预测证据支持,其可靠性最高;CCT II的起始密码子同时有转录、同源和预测证据支持,终止密码子只有表达和预测证据支持,其可靠性次之;CCT III的起始密码子只有表达和预测证据支持,终止密码子同时有表达、同源和预测证据支持,其可靠性再次;CCT IV的起始密码子和终止密码子都只有表达和预测证据支持,其可靠性在4类CCTs中最低。构建基因训练集不仅要考虑基因结构可靠性,而且还要考虑基因的同源性,应尽量避免使用过多同源基因构造训练集,以防止产生训练偏好性。图4给出了根据可靠性和同源性排序生成基因训练集的示意图,由图4可知,BRTGS对4类CCTs进行排序处理的过程为:
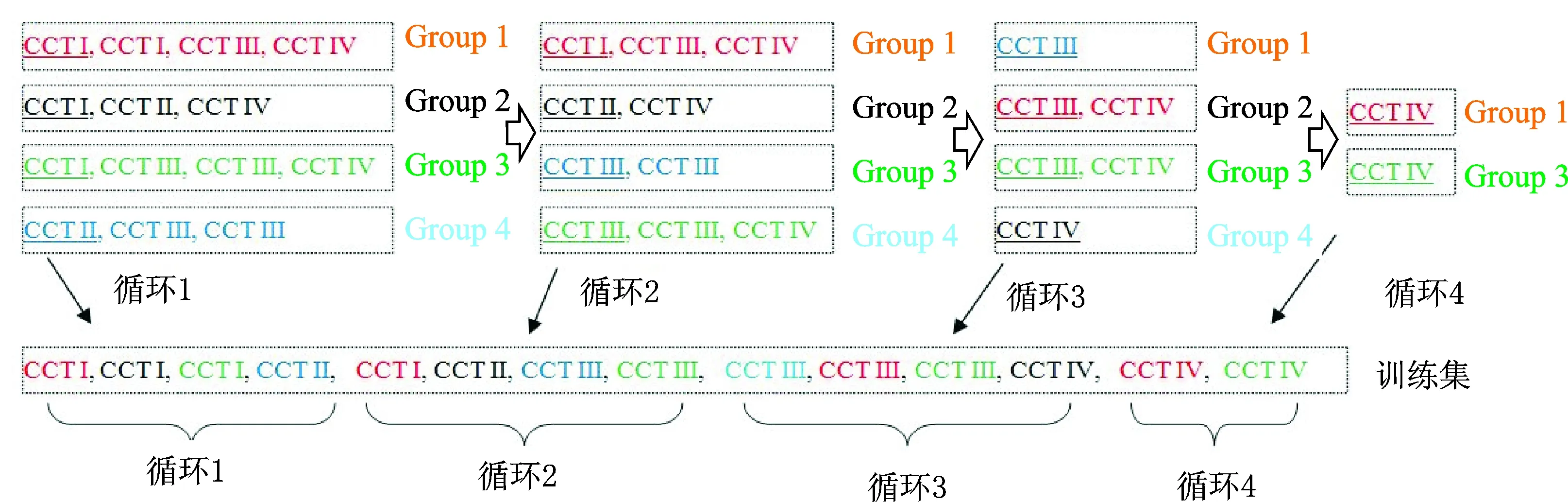
图4 根据可靠性和同源性排序生成基因训练集示意图Fig.4 Demonstration of building training gene set according to reliability and homology
(1)用OrthoMcl[14]对所有的CCT s进行同源聚类,将同源的CCTs聚类到同1个同源分组中,然后对每个同源分组内的CCTs按照“CCT I > CCT II > CCT III > CCT IV”进行可靠性排序,以确保在每个同源分组中高可靠的CCTs被优先选择用于构建训练集。
(2)对同源分组按照包含CCTs类别进行排序。排序后包含CCT I的同源分组将排在最前面,如果同源分组中没有CCT I,那么包含CCT II的同源分组排在最前面,以此类推。如图4所示,在第1轮排序中Group 4不包含CCT I被排到最后;从4个Group中取走第1个CCT后进入第2轮排序,Group 1具有CCT I被排在最前,Group 4和Group 3都没有CCT I和II,所以被排在最后;再次从4个Group中取走第1个CCT后进入第3轮排序,Group 2不包含CCT III被排到最后。
(3)根据步骤(2)对同源分组排序的结果,依次将每个同源分组中的第1个CCT移到训练集中。
(4)重复步骤(2,3),直到每个分组中的CCT都转移到训练集中。
经过上述排序处理后,训练集中的同源基因将被打散。由于训练时,BRTGS只取训练集中前1 000个基因用于训练,因此不仅可以避免同一同源分组中的CCTs被过度采用的现象,而且确保每个同源分组中高可靠性CCTs被优先采用。
3 实验与结果分析
3.1 实验数据
果蝇、拟南芥、人和水稻4个模式物种基因组数据来自于Ensemble(http://asia.ensembl.org),版本号分别为BDGP6.31,TAIR10.31,GRCh38.p2和IGSP-1.0.31。为了模拟不同组装水平的基因组,按照N50定义将每个物种基因组随机打碎。在产生低于N50值的序列时,如果序列长度小于100 bp时,那么这个序列将会被丢弃。在产生大于N50值的序列时,最长序列不超过1 Mb(G640K和G1M的最长序列分别为2 Mb和8 Mb)。4个物种RNA-seq数据都来源于NCBI的SRA(http://www.ncbi.nlm.nih.gov/sra)数据库,访问编号分别为“SRR3091999,SRR3091976,SRR3138705”,“ERR754089,SRR1792926,ERR1109342”,“SRR3184282,SRR3184286,SRR3184279”和“SRR2156305, SRR2047477, SRR2048540”。每个物种的3个RNA-seq数据都分别被依次标为样品1、样品2和样品3。RNA-seq比对和组装分别采用Tophat和Cufflinks实现,参数使用默认值[15-17]。参考蛋白序列来自NCBI-RefSeq数据库,并去除掉来自这4个物种的蛋白序列。
3.2 实验方案
为了评估BRTGS的性能,G2K上最差RNA-seq组装结果和G1M上最佳RNA-seq组装结果(合并了3个RNA-Seq的组装结果)分别用于构建基因训练集。作为比较,核心真核基因定位方法(Core eukaryotic genes mapping approach, CEGMA)(v2.5)、通用单拷贝直系同源基因检测(Benchmarking universal single-copy orthologs, BUSCO)(v1.22)和转录本解码器(TransDecoder)(3.0)也用于构建基因训练集。CEGMA和BUSCO利用保守的核心基因构建训练基因集,因此直接用在G2K和G1M基因组上。TransDecoder会为每个转录本序列报道多个编码区,这里只保留标志为“Complete”且最长的结果作为基因编码结构。本文将对4种方法产生的训练基因数量和质量进行比较。
预测软件的训练效果也是构建基因训练集优劣的重要评价指标。本文用训练集中的前1 000个基因(如果不足,则训练集中的全部基因都用于训练)训练Augustus(v3.2.1),并用其训练后的预测性能评估训练集的训练效果。测试集来自基因组上和训练集不重叠的参考基因。本文将用训练后Augustus在核酸水平、外显子水平以及基因水平上的预测性能评估训练效果,训练和评估过程完全按照Augustus操作手册进行。
3.3 实验结果分析
首先对4种方法构建训练基因数量情况进行比较,结果如表2所示。总体上CEGMA和BUSCO构建的基因数量明显偏少,主要原因是CEGMA和BUSCO构建基因训练集被限制在其定义的真核(或者某个谱系)核心基因集上。这些核心基因数量本来就偏少,在低水平组装的基因组上构建的训练基因则进一步减少。BRTGS和TransDecoder构建训练基因数量偏多,主要原因是BRTGS和TransDecoder构建基因训练集来自于本物种RNA-seq测序数据。RNA-seq测序深度高,其组装结果基本可以覆盖基因组上全部基因(除了少数极低丰度表达或不表达的基因外),因此,构建的训练基因数量明显比CEGMA和BUSCO多。和TransDecoder相比,BRTGS进一步采用蛋白同源证据检验,因此构建的基因数量比较少。尽管如此,BRTGS在每个基因组上仍然构建了超过1 600个基因的训练集。BRTGS构建的训练基因并没有完全来自CCT I,这说明其构建的基因训练集并不完全来自跨物种保守基因。在拟南芥和人的基因组上大约64%和61%来自CCT I,在果蝇和水稻上只有26%和39%来自CCT I。此外,在BRTG构建的基因集中,只有不足5%的基因被同源聚类到一起,因此训练集不具有同源偏好性。

表2 4种方法在不同基因组上构建的训练基因数量
其次,对4种方法构建训练基因质量进行比较,结果如表3所示。以4个物种基因组注释的基因结构为参考,统计4种方法构建的基因训练集的准确性。在外显子水平上,当编码外显子两侧边界和参考基因外显子完全重叠时,该外显子结构被认为正确,否则为错误。在基因水平上,构成基因编码结构的外显子和内含子与参考基因完全一致时,该基因结构被认为正确,否则为错误。准确率为构建训练集中正确外显子(或基因)数与外显子(或基因)总数的比值。在所有测试的基因组上,BRTGS构建的基因结构可靠性最高。BRTGS平均外显子准确性达到93%,分别比CEGMA,BUSCO和TransDecoder高16%,24%和20%;平均基因结构准确性为77%,分别比CEGMA,BUSCO和TransDecoder高40%,40% 和 31%。BRTGS构建的基因结构具有最高的可靠性,主要原因是其利用了表达、同源和预测3个方面证据确保基因结构的完整性和正确性。而CEGMA和BUSCO只利用跨物种保守基因的蛋白同源证据构建基因训练集,难以准确捕捉同源区域外的外显子结构以及起始密码子和终止密码子位置。虽然TransDecoder也利用了RNA-seq组装数据(即RNA-seq表达组装证据),但没有对组装结构正确性以及完整性进行检查,因此基因结构的可靠性也比BRTGS低。
最后,用训练效果(即预测软件训练后的预测性能)对4种方法构建的训练集进行比较,结果如表4所示。在每个测试的基因组上,基于BRTGS基因集训练的Augustus,在核酸、外显子和基因水平上获得的灵敏度和特异度都比其他3种方法高(详细数据未列出)。基因层面的灵敏度和特异度是检查训练效果的最重要指标。用BRTGS基因集训练的Augustus,平均基因灵敏度为33%,分别比CEGMA,BUSCO和TransDecoder高9%,12%和9%;平均基因特异度为22%,分别比CEGMA,BUSCO和TransDecoder高6%,8%和4%。总体上,BRTGS和TransDecoder构建基因集的训练效果比CEGMA和BUSCO好。BRTGS构建的基因训练集具有最好的训练效果主要得益于其充足的训练基因数量和可靠的基因结构。CEGMA和BUSCO构建基因集的训练效果差的原因来自两个方面:(1)训练基因数量偏少、可靠性偏低;(2)在跨物种高度保守的核心基因上训练,难以获得基因组新测序物种自身特性。
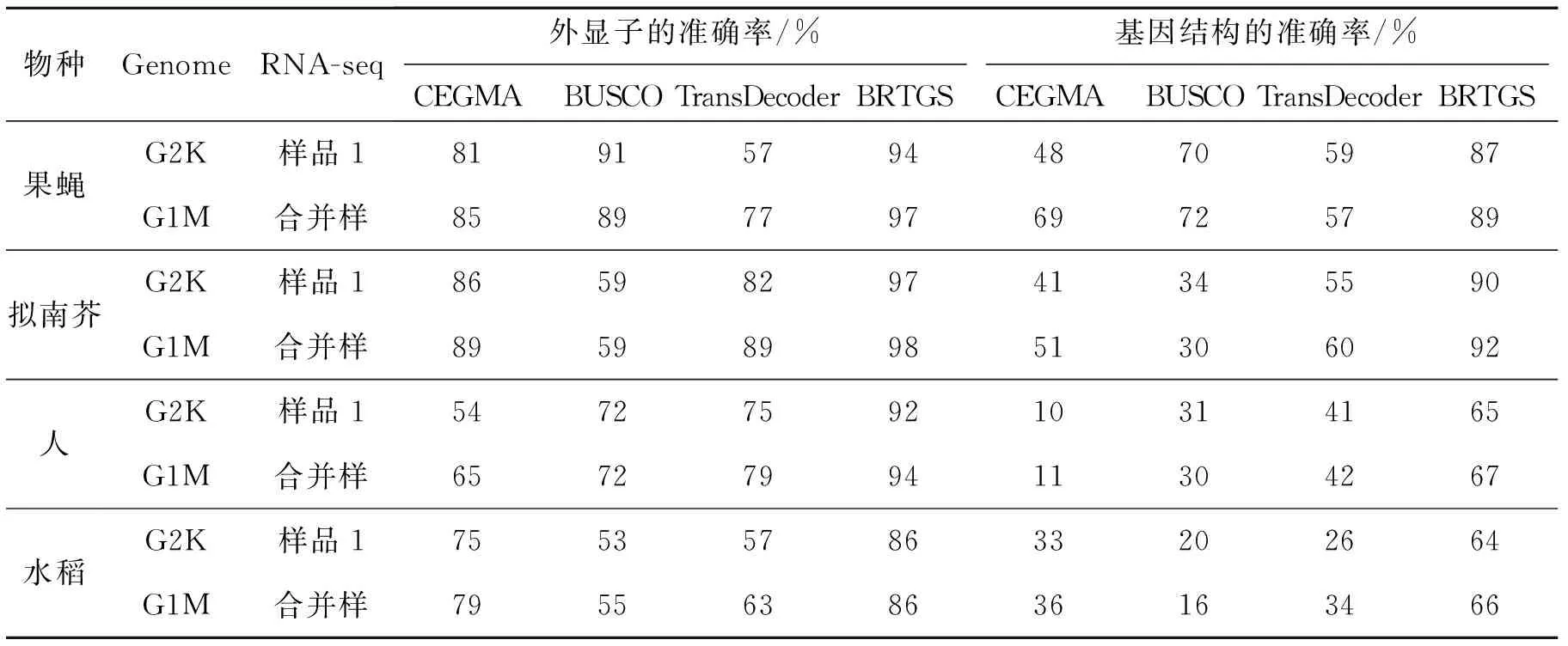
表3 4种方法构建的基因训练集质量

表4 基于4种方法基因训练集的预测软件平均预测性能
4 结束语
长期以来,为从头预测软件准备训练基因集需要专家人工矫正基因结构,这是个耗时、费力的工作。高通量测序技术能够快速获得大量基因表达序列,尤其是三代测序技术的应用能够产生更长甚至全长mRNA序列,这为构造基因训练集进一步提供了十分有价值的序列数据。然而从头预测软件训练不仅需要基因序列数据,而且还要基因元件(如起始密码子、终止密码子、内含子以及外显子等)在基因组上的位置信息。本文提出了一种利用物种自身RNA-seq组装,结合同源证据构建基因训练集的方法,该方法克服了单独利用蛋白同源证据或RNA-seq表达证据构建基因训练集的缺陷,提高了构建基因训练集的质量。通过比较分析,该方法构建的基因训练集不仅具有更高质量的基因结构,而且具有很好的训练效果。值得注意的是,由于基因编码区决定了基因功能,因此本文提出的方法只关注训练基因编码区结构的准确性。事实上,大量研究表明,基因的非编码区(Untranslated region,UTR)也非常重要,比如microRNA经常结合在3UTR上实现对基因的调控。下一步研究重点是进一步优化本文提出的方法,利用RNA-seq测序数据尤其是三代测序长序列数据为基因训练集补充结构完整的UTR。
猜你喜欢
汉字汉语研究(2021年2期)2021-08-30
今日农业(2021年11期)2021-08-13
中西医结合肝病杂志(2020年2期)2020-10-27
生物学通报(2020年11期)2020-10-22
汉字汉语研究(2019年2期)2019-08-27
中成药(2018年7期)2018-08-04
中成药(2018年7期)2018-08-04
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
河北书画研究(2016年3期)2016-04-28
遗传(2014年3期)2014-02-28
