
罐组分级逆流提取技术在娑罗子提取工艺中的应用*
2018-09-05 06:12叶利春刘华侨张晓存石召华
世界科学技术-中医药现代化 2018年4期
石 沁,叶利春,刘华侨,张晓存,吴 灯,石召华**
(1.武汉理工大学化学化工与生命科学学院 武汉 430070;2.武汉爱民制药股份有限公司 鄂州436070;3.湖北省天然组分药物工程技术研究中心 鄂州 436070)
娑罗子为七叶树科植物七叶树Aesculus chinensisBge.、渐江七叶树AesculuschinensisBge.var.chekiangensis(HuetFang)Fang 或 天 师 栗 Aesculus wilsonii Rehd.的干燥成熟种子。秋季果实成熟时采收,除去果皮,晒干或低温干燥。为中国药典收载的中药[1]。娑罗子主要含有七叶皂苷、脂肪油等成分[2-8]。国内外对七叶皂苷的研究报道较多,以婆罗子为原料开发的药用制剂主要有七叶皂苷外用凝胶剂、七叶皂苷口服片剂和七叶皂苷钠注射剂[9],因此提高娑罗子药材中七叶皂苷的提取率对于提高生产效率、降低生产成本、实现绿色生产具有重要意义。
传统中药提取方法主要包括水煎煮法、浸渍法、渗漉法、回流法、溶剂提取法等[10]。随着中药领域的发展,传统中药提取方法已不能满足工业化生产要求。罐组分级逆流提取[11](Multi-stage Countercurrent Extraction,MCCE)是集萃取、重渗漉、动态和逆流技术为一体的具有多种用途的新型中药提取工艺。该提取技术具有工艺简单、效率高、节省溶剂、能耗低等优点,是一种值得推广的新技术、新工艺。本研究运用“动态循环阶段连续逆流提取装置”,以料液比、乙醇浓度、阶段提取时间、提取温度为考察参数,优化最佳提取工艺参数,并在最佳工艺参数下比较罐组分级逆流提取技术和传统提取技术,为七叶皂苷提取工艺技术改造提供依据。
1 仪器与试药
1.1 仪器
高效液相色谱仪(1260,安捷伦);分析天平(AB265-S,梅特勒);集热式恒温加热磁力搅拌器(DF-101S,巩义市予华仪器有限公司);台式低速离心机(TD5A-WS,长沙维尔康湘鹰离心机有限公司);电热恒温鼓风干燥箱(DHG-9245A,上海精宏实验设备有限公司);超高速多功能粉碎机(SZ-2000B-3,永康市善竹贸易有限公司);电子调温电热套(98-1-B,天津泰斯特仪器有限公司)。
图1 对照品色谱图
图2 供试品色谱图
1.2 试药
七叶皂苷钠对照品(100346-201703,中国食品药品检定研究院);娑罗子药材(161203F,武汉爱民制药股份有限公司);色谱乙腈(17065093,天地);分析级乙醇、磷酸(均为国药集团化学试剂有限公司);纯化水(杭州娃哈哈集团有限公司)。
2 试验方法
2.1 七叶皂苷A、七叶皂苷B含量测定
(1)色谱条件与系统适应性试验。
以十八烷基键合硅胶为填充剂,以乙腈-0.2%磷酸水(36∶64)流动相,流速为1 mL·min-1;柱温为30℃,检测波长为220 nm,进样量为10 μL。理论塔板数按七叶皂苷A峰计算不低于5 000,七叶皂苷A、七叶皂苷B峰的分离度不小于1.5。对照品及供试品对应的色谱图见图1、图2。
(2)对照品溶液的制备。
取七叶皂苷钠对照品适量,精密称定,加甲醇制成每1 ml含有1.5 mg的溶液,即得。
(3)供试品溶液的制备。
取娑罗子粉末1.0 g,精密称定,置具塞锥形瓶中,精密加入甲醇50 mL,称定质量,超声提取30 min,放冷,用甲醇补足减失的质量,摇匀,经0.45 μm微孔滤膜滤过,即得。
(4)线性范围考察。
取七叶皂苷钠对照品适量,精密称定,甲醇溶解配成浓度分别为0.804、0.953、1.214、1.436、1.724 mg·mL-1的混合对照品溶液,分别精密吸取10 μl注入高效液相色谱仪,按上述色谱条件进行检测。以峰面积(Y)为纵坐标,以质量浓度(X)为横坐标,绘制标准曲线。七叶皂苷A的线性回归方程为Y=23412.34X-1923.27,R2=0.9996;七叶皂苷B的线性回归方程为Y=24139.25X-1309.63,R2=0.9995。表明七叶皂苷A、七叶皂苷B分别在 0.219~0.587 mg·mL-1、0.236~ 0.489 mg·mL-1的范围内线性关系良好。
(5)精密度考察。
取七叶皂苷钠对照品溶液,按上述色谱条件连续进样6次,测定七叶皂苷A、七叶皂苷B的峰面积,计算相应的RSD值分别为0.09%、0.12%,表明仪器精密度良好。
(6)稳定性试验。
取娑罗子供试品溶液,按上述色谱条件分别于0、4、8、16、24、48 h进样,测定七叶皂苷A、七叶皂苷B的峰面积,计算相应的RSD值分别为1.07%、1.13%。表明样品在48小时内稳定。
(7)重复性试验。
取娑罗子样品粉末约1.0 g,共6份,精密称定,依(3)项下分别制备供试品溶液,按上述色谱条件分别进样,测定七叶皂苷A、七叶皂苷B的峰面积,根据线性回归方程计算含量,所得含量相应的RSD值分别为0.84%、0.69%。表明方法具有良好的重现性。
(8)回收率试验。
取已知含量的娑罗子粉末约1.0 g,共6份,精密称定,分别精密加入七叶皂苷钠对照品约75 mg,按(3)项下方法制备供试品溶液,得到6份供试品溶液。按照上述色谱条件进样,根据峰面积计算含量,计算平均回收率。七叶皂苷A的平均加样回收率为99.82%,RSD为0.66%;七叶皂苷B的平均加样回收率为100.03%,RSD为0.68%。
2.2 出膏率的测定
取娑罗子提取液,滤过,精密量取适量,置于干燥至恒重的蒸发皿中,水浴挥干,于105℃干燥3小时,置干燥器中冷却1小时,迅速称重,按下述公式计算出出膏率(W1为蒸发皿恒重与干膏重之和,W2为蒸发皿恒重,M为药材质量,V1为样品溶液总体积,V2为量取的药液体积)。
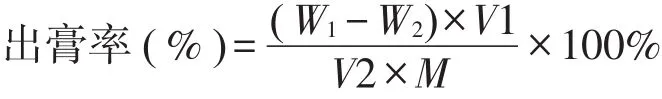
2.3 罐组分级逆流提取工艺[12~15]
(1)罐组分级逆流提取工艺过程。
第1罐的3次提取为梯度形成阶段,即提取3次,每次加入新溶媒,得到提取液A1、A2、A3,其中A1储存。然后开始逆流提取阶段,A2作为第2罐第1次提取的溶媒,得B1,储存;A3作为第2罐第2次提取的溶媒,得B2;B2作为第3罐第1次提取的溶媒,得C1,储存;依次循环提取。每罐最后一次提取,均加入纯溶媒。最后合并A1B1C1C2C3得总提液,其过程见图3所示。测定总提液中七叶皂苷A和B的含量并计算转移率和出膏率,计算综合评分。
(2)正交试验优化提取工艺。
在前期预试验的基础上,选取料液比、乙醇浓度、阶段提取时间、提取温度为考察因素,以七叶皂苷A+B转移率、干膏得率为主要考察指标,按L9(34)表进行正交试验。考察因素与水平见表1,实验安排与结果见表2,方差分析结果见表3。
运用正交设计助手Ⅱv3.1设计正交试验,综合评价七叶皂苷A+B转移率和出膏率2个评价指标对提取工艺的影响,其权重按照8∶2分配,计算综合得分:
综合得分/%=((Xi/Xmax)×80%+(Yi/Ymax)×20%)×100
其中,Xmax、Ymax分别为正交试验结果中的七叶皂苷A+B收率和干膏得率的最大值。
由表2、表3可知各因素对综合评分的影响程度为B乙醇浓度>A料液比>C提取时间>D提取温度,其中,乙醇浓度和料液比对综合评分有显著性影响(P<0.05)。由极值比较可知,四个因素三个水平之间的趋势分别为A3>A2>A1,B3>B2>B1,C1>C3>C2,D2>D3>D1,故优选出的最佳罐组提取工艺为B3A3C1D2,即三罐组条件下药材粗粉加10倍量70%乙醇以60℃每阶段提取20 min。
(3)工艺验证试验。
按照优选出的最佳提取工艺条件,重复试验3次,七叶皂苷A+B转移率和干膏得率结果见表4。由表4可知,3批提取物的综合评分分别为97.73、99.04、99.31(RSD为0.85%),说明罐组分级逆流提取工艺稳定,重现性较好。
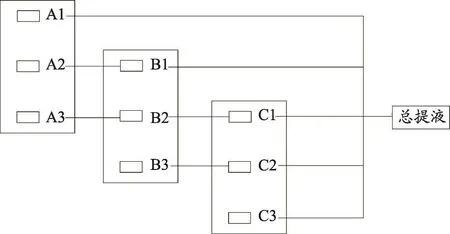
图3 罐组分级逆流提取过程示意图
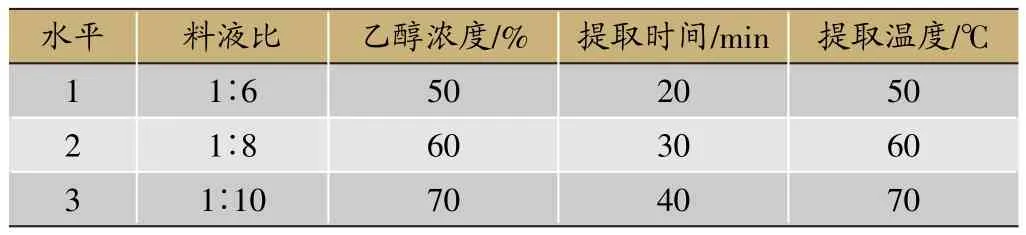
表1 正交试验因素水平表
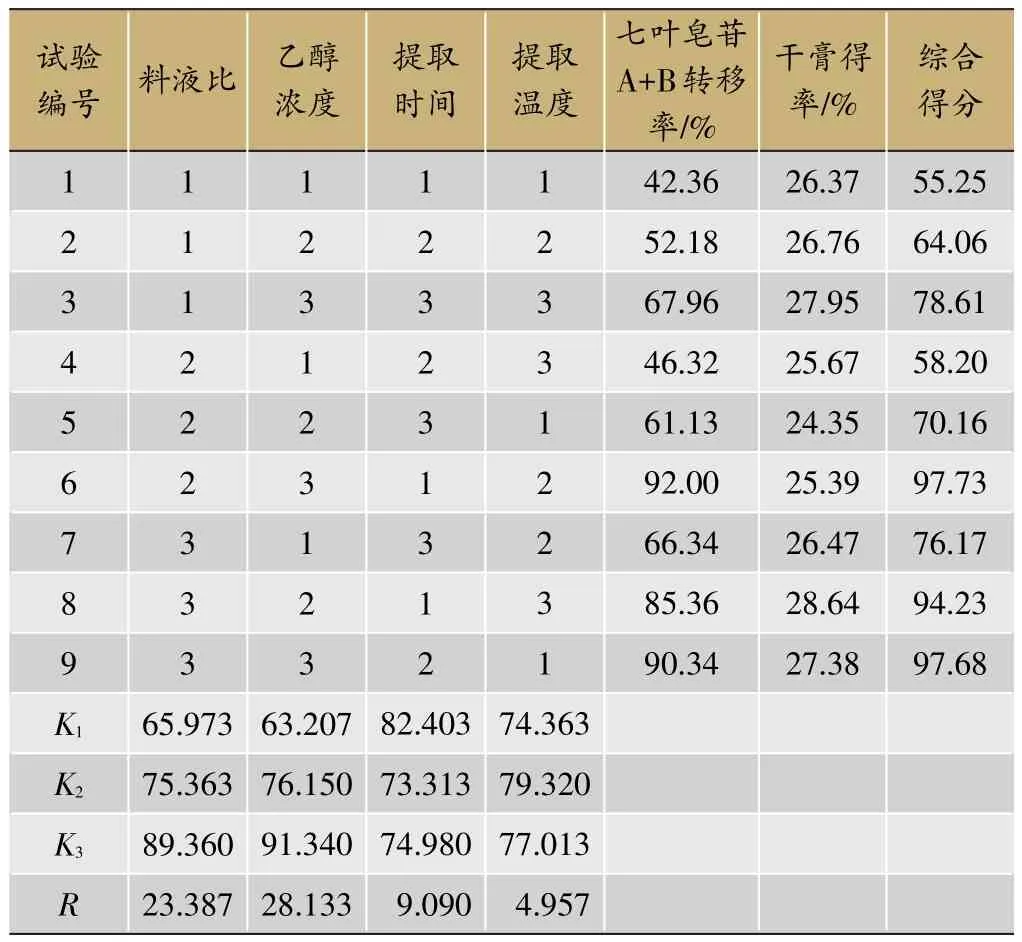
表2 正交试验结果
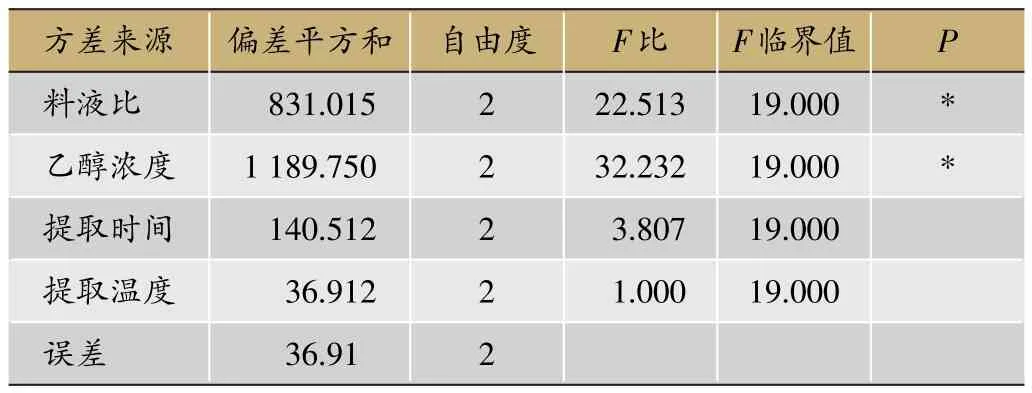
表3 方差分析结果
(4)与传统提取方法的比较试验。取相同量的药材粗粉,按上述优选的罐组分级逆流提取工艺和传统提取工艺(将药材粗粉浸泡2 h,过滤,分别加入10-8-8倍量的乙醇回流3次,前1次回流2 h,后2次各回流1 h)进行比较,两种工艺分别提取3次(n=3),以提取物中七叶皂苷A+B转移率、干膏得率及特征图谱为指标,比较两种提取工艺的优劣。
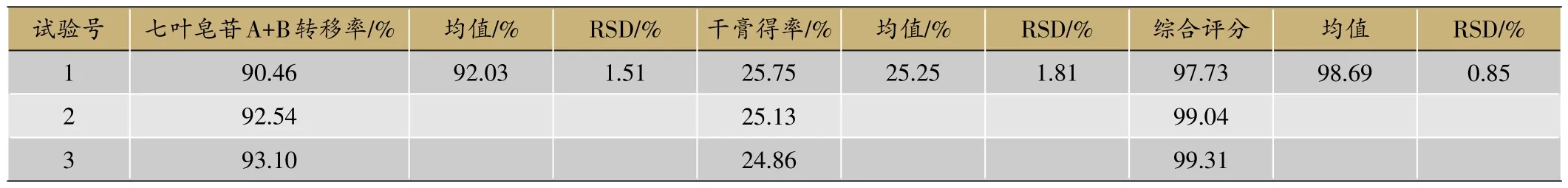
表4 工艺验证试验结果
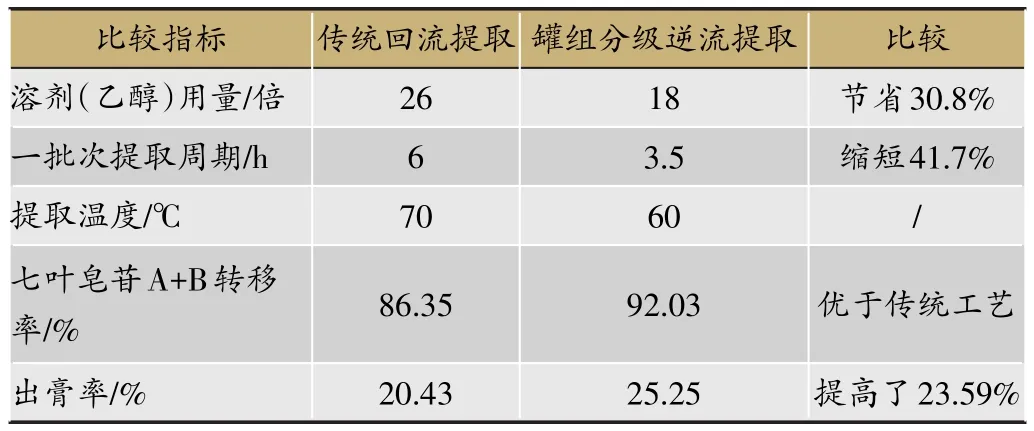
表5 两种提取方法提取效果比较
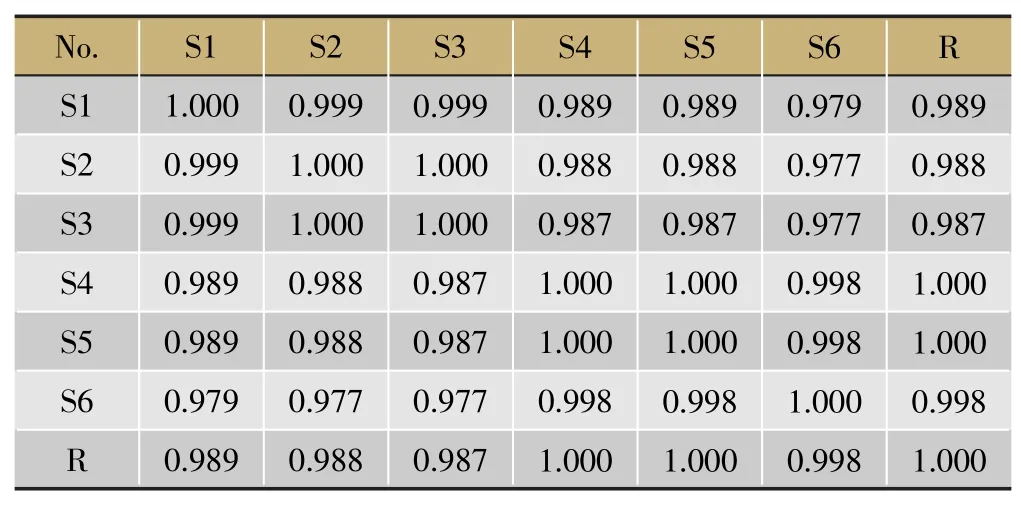
表6 两种提取方法下6批提取物特征图谱相似度评价
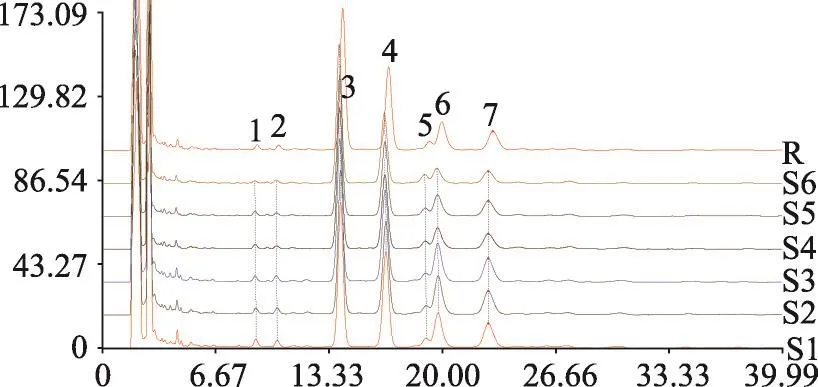
图4 两种提取方法下6批提取物的特征图谱
1)七叶皂苷A+B转移率和干膏得率 试验结果见表5。由表5分析可知,与传统热回流提取方法相比,采用罐组分级逆流提取法在提高有效成分转移率和出膏率的同时降低了提取温度、有效节省溶剂用量,显著缩短提取周期,提高了提取效率,降低了后期浓缩能耗,具有较大的成本优势。
2)特征图谱建立及相似度分析 将两种提取工艺下的6批供试品溶液按2.1.1项下色谱条件测定并建立HPLC指纹图谱,见图4。利用“中药色谱指纹图谱相似度评价系统(2004A)”,分析各样品指纹图谱与对照指纹图谱之间的匹配度及相似性,所得相似度结果见表6,匹配数据见表7。
由表6、表7分析可知,6批特征图谱的相似度均大于0.9,且共有峰个数均相同,表明两种提取方法下所得提取物的化学组成及形态特征高度相似;但是,6批特征图谱中各共有峰的峰面积有较大差异(除峰3外,其余共有峰的RSD均大于5%,n=6),S1-S3各共有峰的峰面积明显大于S4-S6中各共有峰的峰面积,表明罐组分级逆流提取对各组分的提取率高于传统热回流提取。综合分析,与传统热回流提取方法相比,罐组分级逆流提取法在不改变提取成分的基础上能明显提高各个化学成分的提取率。
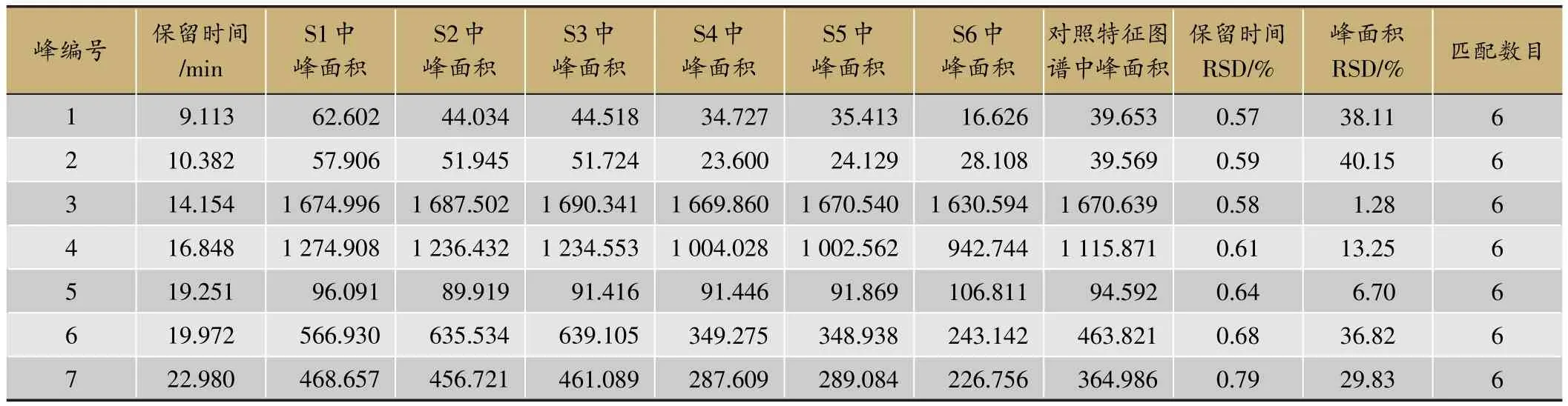
表7 两种提取方法下6批提取物特征图谱匹配数据
3 讨论
由于使用了罐组分级逆流提取工艺,最大程度提高了乙醇利用率,因而乙醇用量从热回流工艺的26倍减少到罐组分级逆流提取工艺的18倍,使得生产过程
猜你喜欢
现代临床医学(2021年1期)2021-01-26
通化师范学院学报(2020年12期)2020-12-21
小学生作文(低年级适用)(2019年4期)2019-04-29
中成药(2018年9期)2018-10-09
散文诗(2017年18期)2018-01-31
中成药(2017年9期)2017-12-19
中成药(2017年6期)2017-06-13
中成药(2017年6期)2017-06-13
中国卫生(2015年6期)2015-01-22
中国药业(2014年16期)2014-05-14
