
β -环糊精衍生物手性固定相的制备及其在毛细管电色谱-质谱中的应用
2018-09-03 01:54李英杰高立娣刘树仁
分析科学学报 2018年3期
李英杰, 赵 楠, 高立娣, 刘树仁, 徐 蕾
(齐齐哈尔大学化学与化学工程学院,黑龙江齐齐哈尔 161006)
β-环糊精(β-CD)具有一定尺寸的立体手性空腔,可以与许多有机无机分子、离子等通过范德华力、静电引力、疏水作用形成主-客体包合物,具备分子识别和选择性结合的能力[1]。通过对β-CD改性不但可以增加其对手性化合物的拆分能力[2],还可以将其应用在毛细管电色谱(CEC)中作为整体柱手性固定相[3]。CEC是在毛细管内壁上键合、涂渍、填充固定相进行电泳的一种方法,具有毛细管电泳与高效液相色谱的双重优势[4]。但近年来,随着样品复杂程度的增加,研究人员对发展高灵敏度检测技术提出了越来越高的要求[5]。毛细管电色谱-质谱(CEC-MS)联用技术不仅具有分离速度快、检测灵敏度高等优点,还能提供待测样品的结构信息,已成为手性分离分析的一种强有力方法[6 - 7]。
手性药物在生物体内与大分子相互识别之后,所产生的代谢、排泄过程以及体现出来的药理、毒理作用都不相同[8]。因此,对手性药物拆分在医药化学领域显得尤为重要。本文采用原位聚合法,以含乙烯基的β-CD(UPA-β-CD)为功能单体,乙二醇二甲基丙烯酸酯(EDMA)为交联剂,制备了新型的环糊精聚合物手性整体柱。优化了制柱条件,以赖氨酸对整体柱的性能进行评价,在CEC-MS模式下成功拆分了盐酸奥昔布宁、盐酸苄丝肼两种手性药物。
1 实验部分
1.1 仪器与试剂
7100毛细管电泳-6230飞行时间质谱联用仪(美国,安捷伦公司);S-4300扫描电子显微镜(日本,日立公司);Nicolet 380傅里叶变换红外光谱仪(美国,热电公司);石英毛细管(75 μm i.d.,365 μm o.d.)(中国锐丰色谱器材公司)。
乙二醇二甲基丙烯酸酯(EDMA)、3-(异丁烯酰氧)丙基三甲基硅烷(KH570)、对甲基苯磺酰氯(上海阿拉丁试剂公司);偶氮二异丁腈(AIBN)和2-丙烯酰胺基-2-甲基-1-丙磺酸(AMPS)(美国Flake公司);β-CD、顺丁烯二酸酐(天津科密欧公司);DL-赖氨酸(北京化学试剂有限公司);盐酸奥昔布宁、盐酸苄丝肼(中国药品生物制品检定所);HAc、NH4Ac、氨水(色谱纯,美国MREDA公司);N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、乙腈、HCl、NaOH、三氯甲烷、乙二胺、丙酮、正十二醇、环己醇均为市售分析纯级。实验用水由GWA-UN5纯水仪(北京普析通用仪器有限公司)制备。
1.2 环糊精衍生物的制备
按文献方法[9]合成6-en-β-CD。称取1.2 g 6-en-β-CD和0.13 g顺丁烯二酸酐,溶于30 mL DMF中,40 ℃下水浴反应4 h。反应结束后,冷却至室温,用三氯甲烷沉淀,再用丙酮充分洗涤,除去未反应的顺丁烯二酸酐和6-en-β-CD后,于真空干燥箱中干燥[10]。反应原理图如图1所示。
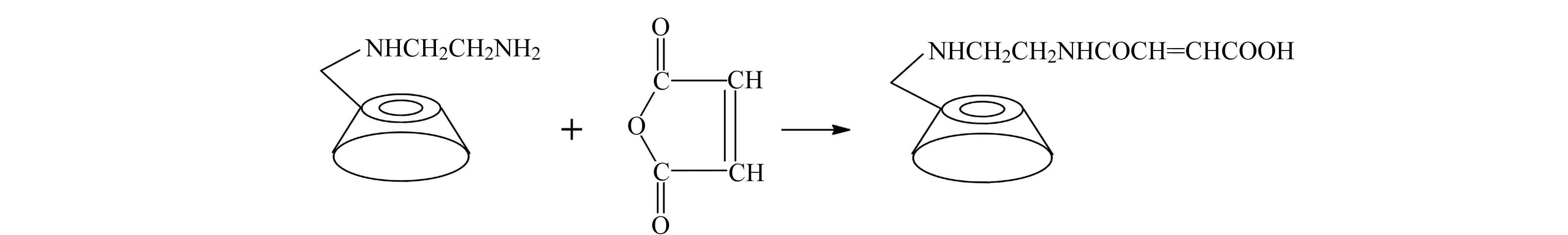
图1 β -环糊精含烯衍生物的制备Fig.1 Schematic diagram showing the preparation of olefin-containing β -CD derivative
1.3 整体柱的制备
将毛细管依次用1 mol/L HCl、水、1 mol/L NaOH溶液、水各冲洗5 min,氮气吹干后注入硅烷化试剂(VKH570∶V丙酮=1∶1 溶液),40 ℃水浴24 h,甲醇冲洗、氮气吹干后备用。分别称取0.025 g UPA-β-CD-DMSO溶液(0.15 g/mL)、0.06 g EDMA、0.15 g正十二醇、0.10 g 环己醇、0.0017 g AIBN 和0.0018 g AMPS 于小瓶中并超声至澄清。取上述液体注入到处理后的毛细管中(填料部分20 cm),两端用橡皮塞封好后,45 ℃水浴反应11 h,反应完成后用甲醇冲洗、氮气吹干。
1.4 样品溶液的配制
分别取0.02 g赖氨酸、盐酸奥昔布宁、盐酸苄丝肼,用水溶解定容至10 mL容量瓶中。配制成2 mg/mL待测样品。使用前经0.22 μm滤膜过滤。
1.5 色谱条件
电色谱条件:20 mmol/L NH4Ac缓冲溶液;运行温度:20 ℃;运行电压:20 kV;进样方式:5 kPa×3 s进样;紫外检测波长:200 nm。
质谱条件:离子源:dual ESI;干燥气温度:325 ℃;干燥气流速:8 L/min;鞘流液组成:50%甲醇-50% 超纯水溶液;流速:0.4 mL/min。
2 结果与讨论
2.1 β -CD衍生物的表征
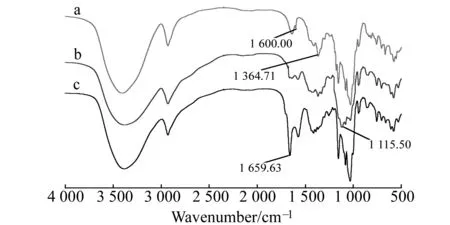
图2 β -CD衍生物的红外(IR)光谱图Fig.2 IR spectra of β -CD derivativesa.6-OTs-β -CD;b.6-en-β -CD;c.UPA-β -CD.
2.1.1红外光谱分析采用红外(IR)光谱对产物进行分析,结果如图2所示。曲线a在1 600 cm-1处为苯环的C=C双键的伸缩振动吸收峰,1 364.71 cm-1处为S=O的对称伸缩振动峰,表明已合成6-OTs-β-CD。曲线b在1 115.50 cm-1处出现了C-N伸缩振动峰,表明化合物已由6-OTs-β-CD转变为6-en-β-CD。曲线c在1 659.63 cm-1处为C=C双键的伸缩振动峰,1 572.68 cm-1处为酰胺基Ⅱ带吸收峰,初步确定合成了目标产物。
2.1.2质谱分析采用质谱对产物(m/z=1 274)进一步验证。在正离子扫描模式下,如图3a所示,m/z1 275.43 为UPA-β-CD的[M+H]+峰,m/z1 297.41为[M+Na]+峰。在负离子扫描模式下,如图3b所示,m/z1 273.41为UPA-β-CD的[M-H]-峰,可以确定合成了目标产物。

图3 UPA-β -CD质谱图 Fig.3 Mass spectra of UPA-β -CD a.positive ion mode;b.negative ion mode.
2.2 整体柱制备条件的优化
在制柱过程中,毛细管内部固定相大孔越多,流动相流经时柱压越低。但对于柱效而言,基质材料中的小孔和中孔数应较多[11]。EDMA和β-CD的比例会影响整体柱对手性药物的拆分效果。EDMA含量增加,材料表面活性位点减小;EDMA含量减小,孔径变大,柱效降低。当EDMA为0.06 g,UPA-β-CD溶液浓度为0.15 g/mL时,制备的整体柱具有良好的分离选择性。温度主要是通过影响AIBN的分解速度来影响毛细管内部孔径分布。反应温度越高,聚合物结合致密,能够承受较高的柱压;反应温度越低,聚合物质地疏松,易被流动相冲掉。综合考虑其机械性能和孔径分布,较好的反应温度为45 ℃。反应时间过短,聚合反应不完全;时间过长,有些官能团可能会遭到破坏,从而影响柱子的分离效果和使用寿命。反应时间为11 h时,整体柱具有较好的通透性和较高的柱效。
2.3 扫描电镜表征
通过扫描电镜(SEM)对所制备的电色谱整体柱内部结构进行表征,从图4可以看出,聚合物成功键合到毛细管内壁上,并且在柱内形成了较密的网状结构,其形态错综复杂,具有较强的机械强度,能承受较大的压力,且整体柱内有较大的孔径,具有良好的通透性。

图4 整体柱固定相扫描电镜(SEM)图Fig.4 SEM image of monolithic column stationary phase
2.4 整体柱固定相红外表征
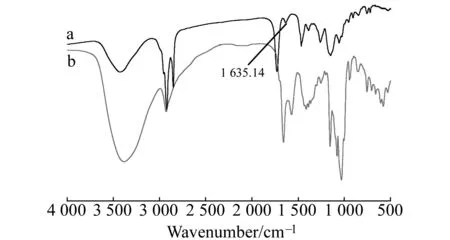
图5 整体柱固定相红外光谱表征Fig.5 Characterization of monolithic column stationary phase with infrared spectrum a.Monolithic column stationary phase;b.UPA-β -CD.
对整体柱固定相进行红外光谱表征,见图5。如图5曲线a所示,在3 422 cm-1处为-OH伸缩振动吸收峰,2 920 cm-1处为-CH2-伸缩振动吸收峰,与图5曲线b的UPA-β-CD相比,1 635.14 cm-1处的C=C峰已经明显减弱,在1 260 cm-1和1 150 cm-1处仍有β-CD的特征峰,说明所制备的整体柱键合上UPA-β-CD。
2.5 柱性能评价
以DL-赖氨酸对整体柱进行性能评价,在20 kV的工作电压下,用20 mmol/L NH4Ac缓冲溶液(pH=5.5)进行连续5次进样,其分离度RS均达到2.0以上。并且其对映体保留时间(tD,tL)的相对标准偏差(RSD)均小于2%,结果表明该整体柱具有良好的分离能力。间歇性使用1个月仍有较好的分离性能,说明整体柱具有较好的稳定性。
2.6 整体柱的应用
用该整体柱对盐酸奥昔布宁、盐酸苄丝肼两种手性药物进行分离,优化CEC分离条件和MS检测条件,两种手性药物可在18 min和22 min内达到基线分离。图6为盐酸奥昔布宁和盐酸苄丝肼对映体的质谱总离子流图(TIC)色谱图,对应的提取质谱图如图7所示。图7a为保留时间为16.91 min的质谱图,m/z358.24是盐酸奥昔布宁的[M-HCl+H]+峰,m/z390.29是盐酸奥昔布宁的[M-HCl+Na]+峰;图7b为保留时间17.49 min的质谱图,m/z358.24是盐酸奥昔布宁的[M-HCl+H]+峰,m/z390.29是盐酸奥昔布宁的[M-HCl+Na]+峰。可以判断图7a和7b为盐酸奥昔布宁的对映体峰。图7c为保留时间19.95 min的质谱图,m/z258.11是盐酸苄丝肼的[M-HCl+H]+峰,m/z290.19是盐酸苄丝肼的[M-HCl+Na]+峰;图7d为保留时间20.91的质谱图,m/z258.12是盐酸苄丝肼的[M-HCl+H]+峰,m/z290.21是盐酸苄丝肼的[M-HCl+Na]+峰。可以判断图7c和7d为盐酸苄丝肼的对映体峰。
图6 两种手性药物的总离子流(TIC)色谱图Fig.6 TIC chromatograms of two chiral drugs
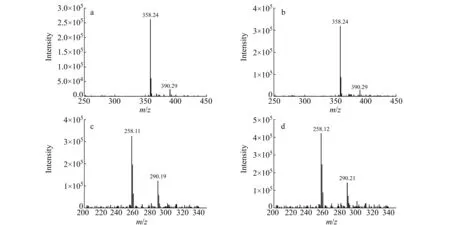
图7 两种手性药物对映体质谱图Fig.7 Mass spectra of the enantiomers of two chiral drugsa,b:MS spectra of oxybutynrn hydrochloride;c,d:MS spectra of benserazide hydrochloride.
2.7 反应机理探讨
盐酸奥昔布宁与盐酸苄丝肼结构具有相似的官能团,与UPA-β-CD作用时苯环可进入环糊精疏水性腔体,形成疏水包合。手性碳原子所连的羟基或氨基基团可与环糊精端口的取代基形成氢键作用。除上述作用外,衍生基团可增强β-CD空腔的柔韧性,使客体分子的手性中心易于接近β-CD的手性部位,有利于增强主客体间的相互诱导作用。
3 结论
本文采用原位聚合法制备了UPA-β-CD毛细管电色谱整体柱,通过赖氨酸对整体柱手性分离性能进行评价,该柱具有较好的手性拆分能力和重现性。在CEC-MS模式下对盐酸奥昔布宁、盐酸苄丝肼两种手性药物进行拆分,分离度达1.76和2.12。此方法试剂消耗量少,对手性药物的分离检测具有一定参考价值。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
云南化工(2020年11期)2021-01-14
现代临床医学(2019年4期)2019-09-10
中成药(2018年8期)2018-08-29
中成药(2018年6期)2018-07-11
中成药(2018年4期)2018-04-26
保健与生活(2018年13期)2018-01-26
中国科技术语(2016年3期)2016-12-04
杂草学报(2015年2期)2016-01-04
中药与临床(2015年5期)2015-12-17
