
加权重基因共表达网络分析识别慢性肾脏病足细胞损伤关键节点基因MAGI2*
2018-09-01 07:19王万鹏唐伯玉沈剑箫李永刚蒯巧林高甄典李娟梁森林陈皓瑜
中国现代医学杂志 2018年24期
王万鹏,唐伯玉,沈剑箫,李永刚,蒯巧林,高甄典,李娟,梁森林,陈皓瑜
(1.江苏省涟水县人民医院,江苏 淮安 225400;2.上海交通大学医学院附属仁济医院,上海 200127)
慢性肾脏病(chronic kidney disease,CKD)与足细胞损伤常常互为因果,足细胞损伤机制极其复杂,多种理化、生物因素均可影响足细胞的功能。本研究拟采用加权基因共表达网络分析(weighted gene coexpression network analysis,WGCNA)挖掘从美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)的基因表达综合数据库(gene expression omnibus,GEO)下载到的足细胞损伤体外模型以及CKD患者肾小球表达谱数据集,分析CKD状态下肾小球足细胞损伤后加权基因共表达网络关系,识别CKD状态下足细胞损伤的相关基因模块及模块内枢纽(hub)基因,为进一步探索CKD足细胞损伤的发生、发展机制提供一种新思路。
1 材料与方法
1.1 数据来源
下载数据集GSE66107、GSE93798,GSE30528、GSE32591和GSE99339,其中GSE66107为嘌呤霉素氨基核苷(purinomycin amino nucleoside,PAN)诱导的足细胞损伤体外模型;GSE93798、GSE30528、GSE32591分别为通过激光微切割获得的IgA肾病、糖尿病肾病(diabetic kidney disease,DKD)、狼疮性肾炎(lupus nephritis,LN)患者肾小球mRNA表达数据,以上4个数据集用于差异表达基因(differentially expressed genes,DEG)分析;数据集GSE99339由多种CKD患者肾小球表达谱数据构成,疾病包括:LN、IgA肾病、膜性肾小球肾炎(membranous glomerulonephritis,MGN)、高血压性肾病(hypertensive nephropathy,HT)、局灶节段性肾小球硬化(focal segmental glomerulosclerosis,FSGS)、肾肿瘤切除(tumor nephrectomy,TN)、微小病变肾病(minimal change disease,MCD)、DKD、 薄 基 膜 病(thin membrane disease,TMD),共133例患者用于WGCNA分析。见表1。芯片的探针注释信息来自Affymetrix公司下载的原始文件。
1.2 数据预处理
采用R语言(3.4.0版)中的Affy包对原始数据进行预处理,包括ReadAffy函数读取原始文件(后缀名为.CEL);全局归一化矫正(Robust Muti-array Average,RMA)函数归一化及对数化;mas5calls函数检测基因表达情况,函数结果表现为“表达(P)”或“缺失(A)”,本研究选择在各数据集样本中表达超过50%的探针用于后续研究;KNN法补充缺失值;limma包进行差异表达值计算,并用贝叶斯方法进行多重检验校正,最终选取阈值错误发现率(false discovery rate,FDR)<0.05及倍数变化(fold change,FC)≥1.5得到DEG。
1.3 WGCNA
1.3.1 模块构建及可视化 GSE99339数据集包含5个批次(bacth),使用sva包中ComBat函数去除批次效应后运行WGCNA包进行基因共表达网络的构建与模块鉴定。共表达网络模块内各基因利用DAVID 6.7(https://david.ncifcrf.gov/)在线工具进行基因本体(gene ontology,GO)分析。利用Cytoscape绘制WGCNA导出的加权共表达基因网络,并通过插件Cluego(v2.3.5)与 Cluepedia(v1.3.5)关联 String 数据库,进行模块内基因GO(功能)和京都基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路富集分析与蛋白间互作网络(protein-protein interactions,PPI)可视化分析。Gluego插件参数设置为pV≤0.05,其余为默认参数。
1.3.2 足细胞特异性阳性标准基因 JU等[1]在过去的研究中使用高通量数据以机器学习为基础发展处一种迭代算法,通过该算法分别定义统计学意义上的50种足细胞特异性标准基因(podocyte standard gene,PSG)。PSG包括:ACTN4、NPHS1及NPHS2等。

表1 数据集纳入及分析结果
2 结果
2.1 足细胞损伤体外模型及CKD患者肾小球差异表达基因分析
经过预处理并删除低表达探针(表达率<50%数据集样本数)后剩余探针分别为26 357、11 118、11 296及7 194个,合并重复探针和删除无注释探针后,最终分别得到6 154、3 882、1 422、840个DEG(FDR<0.05,FC≥1.5)(见表1)。使用R语言ggplot包绘制DEG热图,可见所得DEG可以区分出大部分CKD患者或者损伤的足细胞(见图1A~D)。本研究4个数据集共得到9 217个DEG,共同DEG(coDEG)15个,其中上调基因4个(见图1E),下调基因11个(见图1F),分别为CDKN1C,MYO5C,C1orf21,KBTBD11,NR3C2,FZD2,MAGI2,LPL,ZFPM2,USP46,PRKAR2B,TRIM38,DESI1,CTSS,ITGB2。
2.2 WGCNA构建基因共表达模块
9 217 个DEG中有1 260个DEG表达趋势在2个或2个以上数据集中存在相反(与对照组比较),删除后剩余7 957个DEG,其中4 031个DEG存在于GSE99339预处理后的表达谱中,被用于WGCNA分析(见表1)。网络模块分析结果易受离群样本的影响,因此在构建网络之前去除离群的样本数据显得尤为重要。相关研究常采用芯片间相关度(inter.array correlation,IAC)来评估芯片数据的分布情况。IAC可以通过计算任意一对芯片上所有探针表达水平的相关系数获得,芯片之间的关系可以用树形图来展示,平均IAC值较低的样本,或者在树形图上无法聚类的样本即为离群样本,应被去除。本研究使用WGCNA包中的函数dist()计算样本间的IAC,并通过hclust()函数绘制133个样本4 031个DEG构成的树形图,结果未发现明显离群值和疾病聚集(见图2A);根据动态分层剪切树算法最终得到12个基因模块(见图2B),以颜色的英文命名,其中grey模块包含518个DEG,表示未纳入任何模块的基因集合。
2.3 足细胞损伤相关基因模块鉴定及分析
12个基因模块与未归类的grey模块(见表2),本研究中共包含PSG 22个,其中尤以green和red模块最多,分别为10和4个。同时通过DAVID进行模块内基因功能富集发现green和red模块所含基因与肾小球发育、细胞外基质解聚、细胞机械刺激等足细胞常见的病理状态密切相关,因此笔者认为这两个模块与足细胞损伤关系较为密切。另一方面,与两个模块比较,发现green模块中包含更多的coDEG,因此可以推测green模块中的基因可能参与多种CKD发展中 的足细胞损伤。

图1 各数据集差异基因表达热图及韦恩图
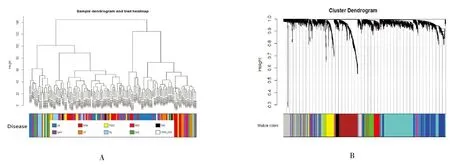
图2 WGCNA分析构建基因共表达模块
2.4 足细胞损伤相关模块加权网络分析
hub基因是指在模块中连接度最高的一系列基因,与全局网络中的枢纽基因比较,模块中的枢纽基因往往更有生物学意义。本文定义的WGCNAhub基因为模块内平均连接度(Kwithin)前5%的基因[2]。结果显示,green模块中MAGI2基因可以同时作为即是green模块的hub基因也是本研究中的coDEG和PSG(见表2),结果高度提示MAGI2不仅具有CKD足细胞损伤的分子标志物潜在价值,也可能在早期足细胞损伤中起到重要生物学作用。选择关联权重阈值≥0.05构建mRNA共表达网络(见图4A),并使用Cytoscape及ClueGO插件并联合String数据库构建green模块内所有基因的KEGG/GO/PPI互作网络,进一步挖掘模块内基因参与的生物学进程及蛋白相互作用,结果显示与MAGI2表达或蛋白互作高度相关的多种基因已经被报道参与足细胞损伤,包括NPHS2、FGF1、BMP7等(见图4B)。

表2 构建出的模块资料及所含基因总结

图3 各模块功能富集结果(取P值最小前3项)

图4 green模块内基因功能及相互作用网络可视化
3 讨论
CKD时足细胞损害机制极其复杂,涉及多种通路及细胞因子[3]。但传统的生物学研究以单基因或蛋白为出发点,因此其仅能对生物系统的局部作出解释,难以对系统的整体进行全面的探索。相比之下,WGCNA是一种从高通量数据中挖掘模块(module)信息的算法。在本方法中module被定义为一组具有类似表达谱的基因,如果某些基因在一个生理过程或不同组织中总是具有相类似的表达变化,那么有理由认为该基因在功能上是相关的,可以把其定义为一个模块(module)。这似乎有点类似于进行聚类分析所得结果,但不同的是,WGCNA的聚类准则具有生物学意义,而非常规的聚类方法(如利用数据间的几何距离),因此该方法所得结果具有更高的可信度,更有利于从生物功能整体入手考虑基因功能以及内在关联[4]。
本研究所纳入的GSE66107数据集是PAN诱导的足细胞损伤的经典体外模型,PAN是嘌呤霉素的衍生物,具有选择性的肾损害作用,PAN所致的肾脏损伤与人类肾病损伤一致,早期表现为足细胞足突融合和消失的微小病变(MCD),随着PAN作用时间的延长和蓄积量的增加,足细胞凋亡增加,进一步发展为局灶节段性肾小球硬化(FSGS),而PAN作用的靶细胞即为足细胞。因此通过足细胞体外损伤模型的基因表达变化与另3个CKD患者肾小球基因变化取交集,有利于排除肾脏其他实质细胞(如系膜细胞和内皮细胞)表达变化的干扰,可以更明确探讨CKD时足细胞内可能存在的基因表达异常。最终将4个数据集所得到的DEG结合含有133个CKD样本表达谱数据进行WGCNA分析,并通过GO富集分析现red和green模块在GO分类上与多种足细胞损伤相关进程较为密切:如机械刺激反应(cell response to mechanical stimulus)[5]、内质网应激反应(response to endoplasmic reticulum stress)[6-7]、肾小球发育(glomerulus development)、细胞外基质分解[8](extracellular matrix disassembly)等。更有价值的是,笔者发现green模块中的hub基因MAGI2也同为本研究中的coDEG和PSG,同时,网络可视化结果显示MAGI2与多种文献已报道的在足细胞损伤或肾脏病中起到重要“著名”基因相关量,如FGF1[9]、BMP7等[10]。这意味着MAGI2可能具有作为足细胞损伤早期分子标志物的潜在价值,并且在足细胞损伤中起到重要的调控作用,同时通过共表达网络的构建识别其高度相关的节点基因,对后续MAGI2的调控机制研究提供重要线索。
本研究不足之处在于从GEO数据库中下载的基因表达谱无法得到相应的临床资料,如肾小球滤过率(eGFR)、血肌酐(Scr)、尿蛋白等,如果可以加入该临床参数作为参考,那么WGCNA分析时更可以将所得模块与该临床信息进行相关性分析,挖掘模块可能所具有的生物学意义,发挥WGCNA的更大作用。
综上所述,本研究通过运用一系列合理的生物信息学手段,分析CKD患者肾小球内与足细胞损伤相关度的15个差异变化基因,同时通过共表达分析构建出足细胞损伤相关基因模块green,研究结果对于CKD,尤其是足细胞损伤的早期诊断提供线索,同时为足细胞保护相关研究提供依据及参考,是WGCNA在CKD足细胞损伤中的一次新型尝试。
猜你喜欢
安徽农业科学(2022年1期)2022-02-14
世界科学技术-中医药现代化(2021年8期)2021-12-21
天津医科大学学报(2021年4期)2021-08-21
江苏农业学报(2021年2期)2021-06-30
昆明医科大学学报(2020年12期)2021-01-26
装备制造技术(2020年1期)2020-12-25
北京航空航天大学学报(2017年2期)2017-11-24
湖北农业科学(2017年10期)2017-06-22
医学研究杂志(2015年9期)2015-07-01
兵工学报(2012年8期)2012-02-23
