
结核病新抗生素靶标和抑制剂研发
2018-08-28 08:06:56冀磊谢建平
生物工程学报 2018年8期
冀磊,谢建平
谢建平 研究员,西南大学生命科学学院现代生物医药研究所副所长,博导,中国微生物学理事/基础微生物/分子微生物与生物工程专委会副主任、中国微生物学会教学工作委员会委员、《遗传》副主编、《生物工程学报》、《药学学报》、《微生物学报》、《中国抗生素杂志》和编委,享受国务院政府特殊津贴专家 (2010),教育部新世纪优秀人才计划入选者 (2011),重庆市缙云英才入选者。主要围绕结核病持续开展研究,先后主持国家传染病科技重大专项和国家自然科学基金等20余项,近5年以通讯作者发表SCI论文80余篇。

结核病新抗生素靶标和抑制剂研发
冀磊1,谢建平2
1 杭州医学院 基础医学部,浙江 杭州 310053 2 西南大学 生命科学学院 现代生物医药研究所 三峡库区生态环境与生物资源省部共建国家重点实验室培育基地,重庆 400715

冀磊, 谢建平. 结核病新抗生素靶标和抑制剂研发. 生物工程学报, 2018, 34(8): 1326–1337.Ji L, Xie JP. Progress in role of novel anti-tuberculosis drug targets and their inhibitors. Chin J Biotech, 2018, 34(8): 1326–1337.
随着结核病耐药现象越来越严重,迫切需要开发新型抗结核药物,而大部分的药物开发主要依赖于新型的抗生素靶标的发现。文中综述了2016年以来新出现的具有开发为抗结核药物潜力的新化合物,并重点阐述了这些新化合物所针对的抗生素靶标,将有助于针对这些靶标进一步开发新型抗结核药物。
抑制剂,抗生素靶标,结核病
结核病 (Tuberculosis, TB) 是严重的全球性公共卫生问题之一,世界卫生组织最新统计显示其危害被严重低估了,目前其致死人口已超越艾滋病,成为全球最为严重的传染病[1]。2016年,全球结核病患者超过1 000万,其与艾滋病的共感染人口占10%[1]。耐多药TB (Multidrug-resistant tuberculosis, MDR-TB) 和广泛耐药TB (Extensively drug-resistant tuberculosis, XDR-TB) 患者数量逐年上升,仅2016年,便新增49万MDR-TB患者[1],成为结核病防控工作的三大挑战之一,遏制耐药TB工作迫在眉睫[2]。
在度过了漫长的没有新型抗结核药物的50年后,通过科研工作者的不懈努力,终于迎来了两种新型抗结核药物贝达喹啉 (Bedaquiline) 和迪拉马尼 (Delamanid)[3]的面世,但这对于目前结核病的严峻形势还是远远不够,急切需要研究新的药物靶标,开发新型的抗结核药物。最近有一些新型的抗结核药物靶标和药物处于不同的研究阶段,这为抗结核工作带来了新的希望。
1 抗结核新靶标研究进展
1998年结核分枝杆菌 (, Mtb) H37Rv全基因组测序的完成,促进了Mtb致病机理的研究,推动了抗结核新靶标的发现[4]。
药物靶标的发现主要是通过遗传方法和功能基因组学方法来实现的。在过去一段时间,多个研究机构通过上述方法,构建了多种新颖的化合物筛选模型;同时利用计算机辅助设计、活性化合物结构改造等手段建立新型化合物库,最终将筛选模型与化合物库有机结合,筛选出十多个新的抑制剂。它们在抗结核药物的开发方面给人们提供了更多的选择。
2 结核病新抗生素靶标及其抑制剂
2.1 细胞分裂蛋白Wag31
细胞分裂蛋白Wag31 (或称为抗原84, Ag84),其在革兰氏阳性菌中的同系物也称为DivIVA[5]。Wag31是Mtb生长中的必需蛋白,该蛋白功能的缺失会导致菌体的死亡[6]。目前的研究认为它没有催化活性,定位于细菌两端,在菌体的生长繁殖中发挥重要的作用[7],是协调菌体细胞延伸的骨架蛋白,与细胞的分裂密切相关[8]。Wag31蛋白高度保守的N-端有1个脂质结合区 (Lipid binding domain, LBD),该基因在枯草芽孢杆菌中的同源基因主要负责极性膜定位[9]。在丝氨酸/苏氨酸蛋白激酶 (Ser/Thr protein kinases, STPKs) PknA和PknB的共同催化下,Wag31蛋白73位苏氨酸被磷酸化,从而激活Wag31蛋白功能[10]。Wag31蛋白功能的异常,会导致菌体形态改变,产生不对称的膨胀,最终使菌体一端变圆[7]。该蛋白编码基因在Mtb中编号为Rv2145c,共783 bp,编码260个氨基酸,其中第73位为苏氨酸。
Singh等[8]在其研究中发现了3种有抑制Mtb生长效果的化合物:AminoPYrimidine-Sulfonamide (APYS1)、APYS2和APYS3 (图1)。在链霉素突变株18b注射的小鼠模型中[11],发现3种化合物对于非复制期的Mtb没有任何杀菌作用,而在Mtb的复制期可以有效抑制其生长,提示这3种化合物可能作用于细胞生长中的关键步骤。同时还发现,在RAW巨噬细胞中加入浓度为20 mmol/L的APYS1或在成纤维细胞中加入浓度为100 mmol/L的APYS1,APYS1均未表现出抗Mtb H37Rv活性。其中最有潜力开发为抗结核药物的APYS1,其最小杀菌浓度 (Minimum bactericidal concentration, MBC)为0.6 μmol/L,最小抑菌浓度 (Minimal inhibitory concentration, MIC) 为0.3–0.6 μmol/L。
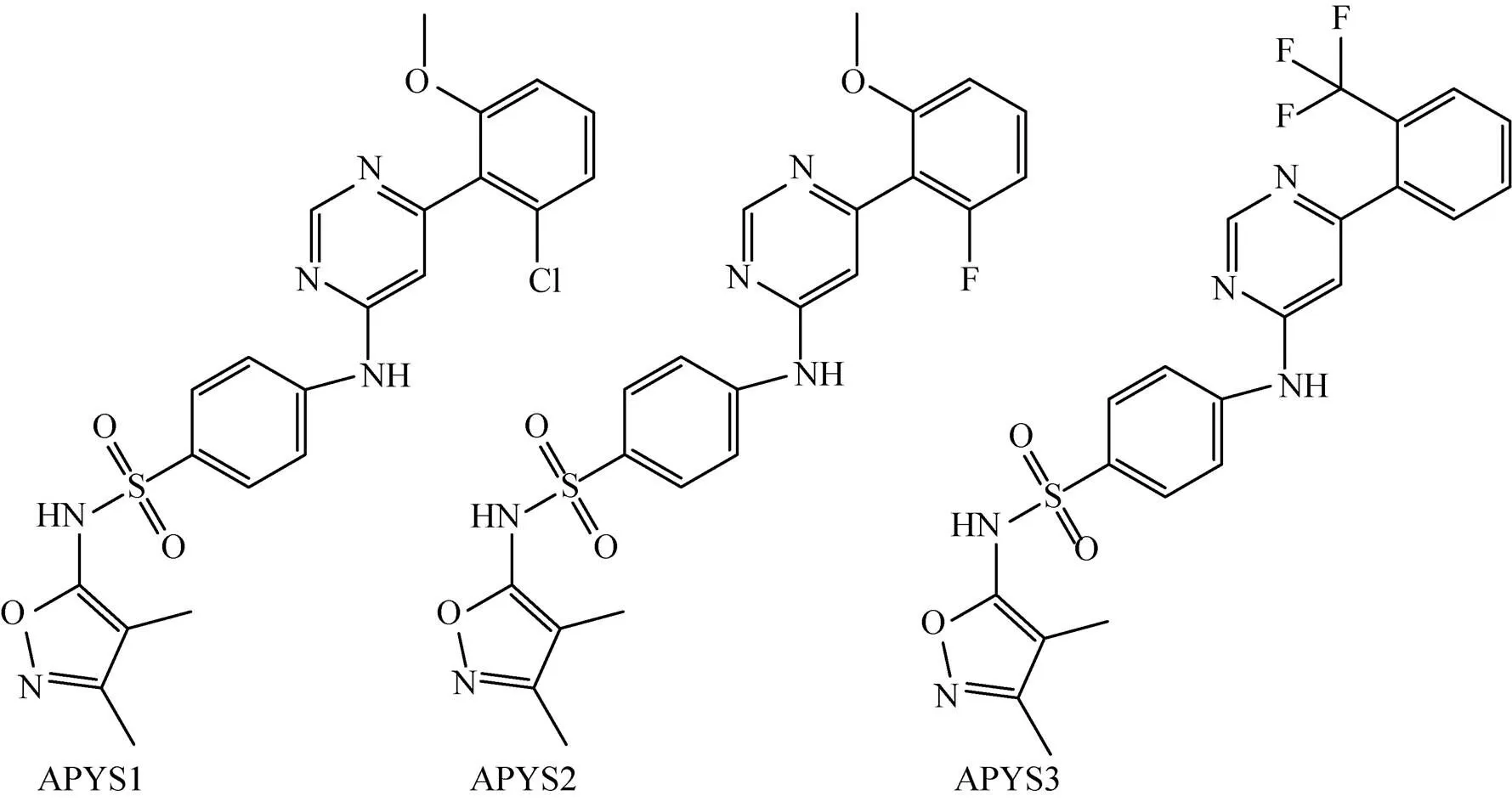
图1 通过表型筛选出的抗结核化合物APYS1、APYS2和APYS3
尽管APYS1的抑菌效果十分明显,但其作用机制还未得到揭示。Singh等[8]发现APYS1在基因发生突变的Mtb中丧失了抗菌活性,推测其作用位点为Wag31蛋白。但其后续实验发现该化合物并未直接与Wag31相互作用,推测其进入胞内后,结合一种目前还未知的蛋白形成蛋白复合物,该蛋白复合物与Wag31相互作用后导致菌体死亡。但该化合物是否通过该机制发挥作用,还有待实验进一步验证。
2.2 GlmU双功能酶
GlmU蛋白为双功能的酶类,同时具有乙酰转移酶和尿苷酰转移酶两种酶的活性。其C-端发挥乙酰转移酶活性,催化葡萄糖胺-1-磷酸(Glucosamine-1-phosphate, GlcN-1-P) 和乙酰辅酶A合成N-乙酰葡萄糖胺-1-磷酸 (N-acetyl-glucosamine- 1-phosphate, GlcNAc-1-P) 和辅酶A,而其N-端发挥尿苷酰转移酶功能,进一步催化GlcNAc-1-P和UTP合成UDP-N-乙酰氨基葡糖 (UDP-N-acetyl- glucosamine, UDP-GlcNAc)[12-13]。在微生物和哺乳动物中均普遍存在尿苷酰转移酶活性,但Mtb乙酰转移酶活性仅存在于微生物中[14],所以针对该酶的抑制剂可以作为潜在的抗结核药物来研究。通过两步催化获得的UDP-GlcNAc是合成肽聚糖和脂多糖的底物,而后两者是Mtb细胞壁的主要成分[14-15]。该酶功能的缺失将会直接影响Mtb的生存。该蛋白编码基因在Mtb中编号为Rv1018c,共1 488 bp,编码495个氨基酸。
Mehra等[16]利用计算机辅助设计手段,从容量为20 000的化合物库中,筛选出了15个针对GlmU蛋白抑制率超过40%的化合物 (表1)。而其中化合物5810599的抑制率高达82.5%,通过剂量依赖性实验评估出该化合物的半抑制浓度 (Half maximal inhibitory concentration, IC50) 为 (9.018± 0.04) μmol/L。具有开发为以GlmU蛋白为靶标新型抗结核药物的潜力。

表1 体外实验中用100 μmol/L药物处理菌体后抑制率大于40%的化合物
2.3 MmpL3转运蛋白
分枝杆菌膜蛋白 (Mycobacterial membrane protein large, MmpL) 家族成员是一种多底物外排泵,属于跨膜转运蛋白抗性/结节/细胞分裂 (Resistance-nodulation-cell division, RND) 透明质酸酶超家族[17]。底物的跨膜转运受到跨膜质子电化学梯度的质子动力势 (Proton motive force, PMF) 驱动。MmpL蛋白不仅介导底物的跨膜运输,其在结核和非结核分枝杆菌的药物外排中也发挥举足轻重的作用,可将胞质中的抗生素排到胞外,从而导致菌体的耐药。Mtb中共存在13个MmpL蛋白,只有MmpL3蛋白的缺失会导致其无法在体外生存[18]。MmpL3蛋白主要负责海藻糖单霉菌酸酯 (Trehalose monomycolate, TMM) 向外转运[19],同时介导亚铁血红素的胞内转运[20-21]。
该蛋白编码基因在Mtb中编号为Rv0206c,共2 835 bp,编码944个氨基酸。处于临床前研究的二芳基吡咯衍生物BM212,可以有效抑制Mtb生长,其MIC=0.7–1.5 μg/mL[22],其通过抑制MmpL转运分枝菌酸进一步影响Mtb细胞壁的合成[23]。Ramesh等[24]在其研究中合成了BM212的结构类似物 (图2),该化合物主要针对的也是MmpL蛋白,其MIC=0.031 μg/mL,活性比BM212高 25倍,具有很好的抑菌效果。
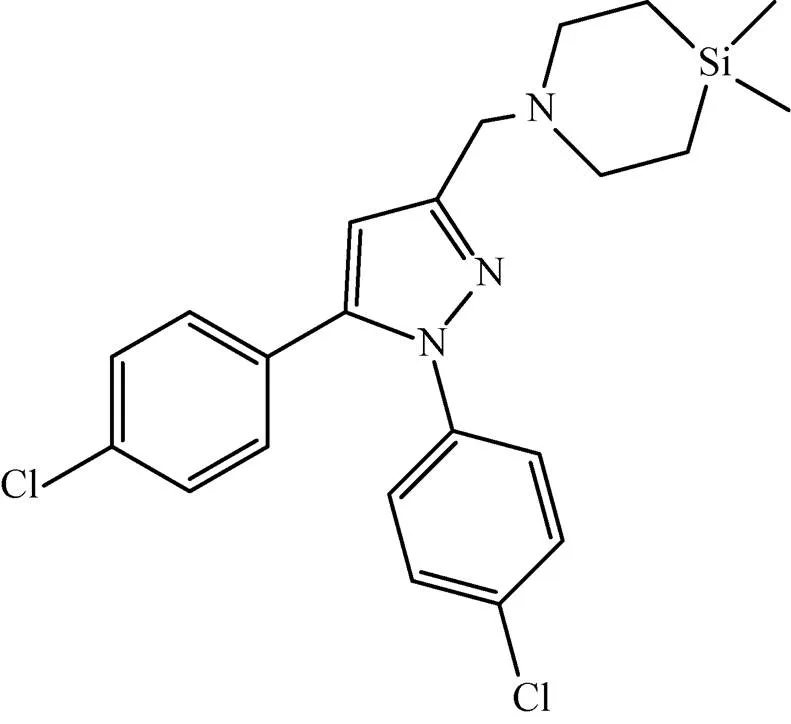
图2 化合物18a
2.4 Mtb蛋白酪氨酸磷酸酶 (Protein tyrosine phosphatase, Mptp)
各种蛋白酪氨酸磷酸酶 (Protein tyrosine phosphatases, PTPs) 共同构成一个大的信号酶类家族,其与蛋白酪氨酸激酶家族 (Protein tyrosine kinases, PTKs) 共同调节细胞水平的蛋白酪氨酸磷酸化[25-26]。PTPs和PTKs中任何一个酶功能的缺失,都会导致蛋白酪氨酸磷酸化的异常,进一步导致人类罹患癌症、糖尿病或免疫功能障碍等疾病[27]。有些病原微生物的感染也可以导致PTPs和PTKs的功能异常。Mtb中MPtpA和MPtpB对于其在巨噬细胞和各种动物感染模型中的存活也发挥举足轻重的作用[28-33]。尽管Mtb本身缺乏内源性蛋白酪氨酸磷酸化作用,但MPtpA和MPtpB可通过与巨噬细胞的相互作用,调节菌体与巨噬细胞间的平衡,保证菌体在巨噬细胞中的生存。MPtpA可将其底物液泡蛋白质排序蛋白33b (Vacuolar protein sorting protein 33b, VPS33B) 去磷酸化,阻止吞噬溶酶体发挥作用,从而导致膜泡中巨噬细胞液泡型ATP酶 (Vacuolar-H+-ATPase, V-ATPase) 的清除[34-35]。一旦进入巨噬细胞,MPtpB随即活化Akt信号肽,同时阻止ERK1/2和p38的活化,直接导致巨噬细胞的凋亡和信号分子的释放。一旦将MPtpA和MPtpB功能缺失,Mtb在干扰素γ (Interferon-γ, IFN-γ) 诱导的巨噬细胞中的存活率和豚鼠结核慢性感染模型中的载量均会发生显著下降[28,36]。MPtp功能缺失的Mtb感染豚鼠模型后可以使其对野生型Mtb的抵抗力增强[33],提示我们针对MPtp的抑制剂可以作为新型的抗结核药物。该酶可调控细胞增殖, 在信号转导途径中发挥调控因子的作用。
MPtpA蛋白编码基因在Mtb中编号为Rv2234,共492 bp,编码163个氨基酸。而MPtpB蛋白编码基因在Mtb中编号为Rv0153c,共831 bp,编码176个氨基酸。
陈冬妮等[37]从海洋真菌的代谢产物中分离出多种卷线孢菌素的衍生物 (图3),通过建立以MPtpB为靶点的筛选模型,从卷线孢菌素的衍生物库中筛选出了化合物25,该化合物的IC50为 (64.6±9.1) µmol/L,远低于卷线孢菌素 (IC50为 (327.6±60.4)µmol/L),具有开发为新型抗结核药物的潜在价值。
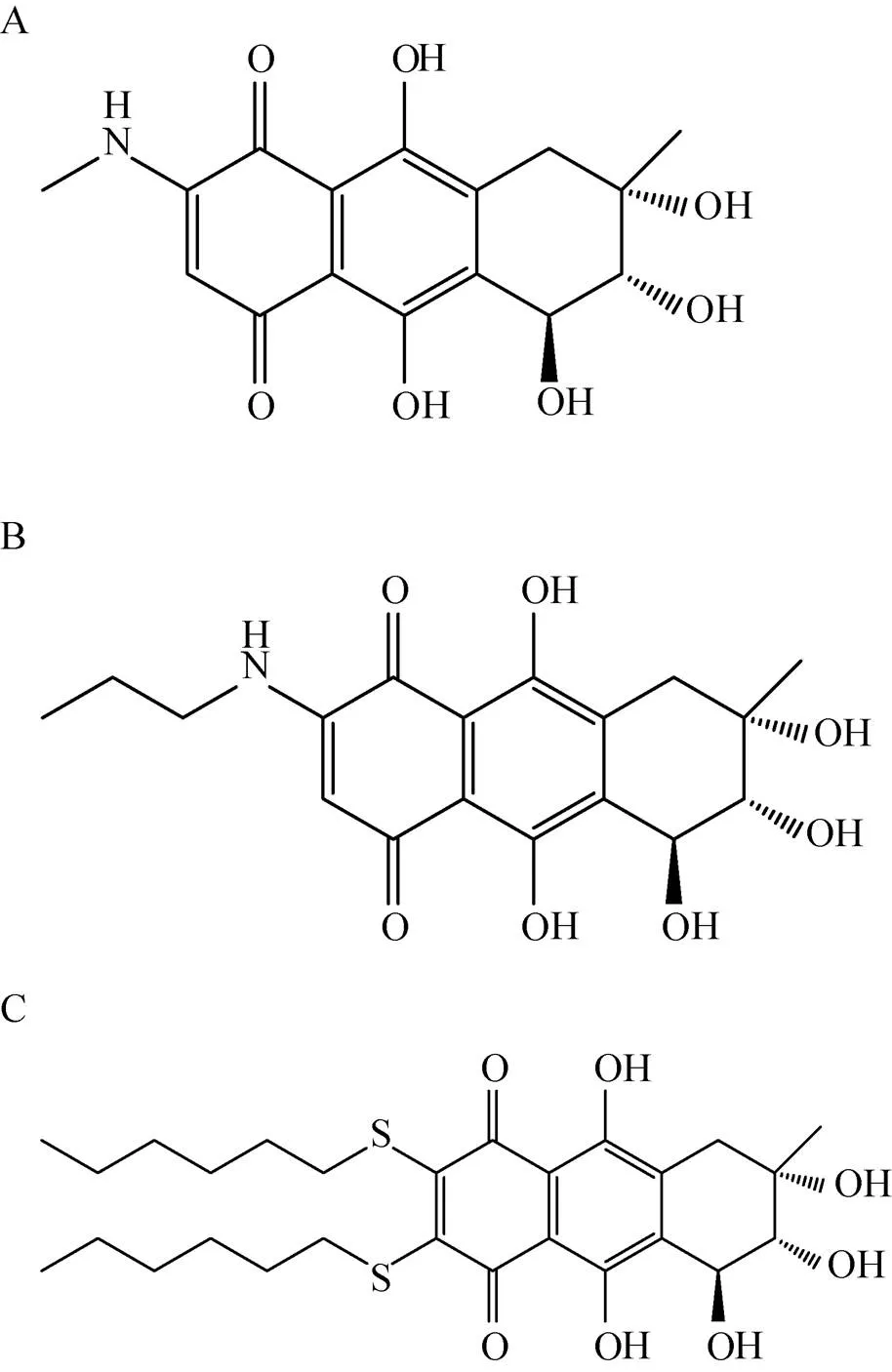
图3 卷线孢菌素衍生物
2.5 磷酸-N-乙酰胞壁酸-五肽转位酶 (Phospho- N-acetylmuramoyl-pentapeptide-transferase, MraY)
肽聚糖是细菌细胞壁的主要组成成分,其生物合成中的任何关键步骤受到影响,均可以导致菌体的死亡,所以这些关键步骤的酶类均可以作为药物靶标进行研究。大肠杆菌中肽聚糖的合成过程已经研究得较为明确[38-40],Mtb中也存在类似的肽聚糖合成途径,但是与大肠杆菌存在某些差别:1) 胞壁酸中除了N-乙酰基 (N-acetyl, NAc) 基团外,增加了N-羟乙酰基 (N-glycolyl, NGlyc);2) 肽聚糖多肽部分的羧酸类被酰胺化;3) 更多的甘氨酸或丝氨酸残基[41-43],而Mtb中的胞壁酸绝大部分均发生了N-羟乙酰化[41]。
磷酸-N-乙酰胞壁酸-五肽转位酶普遍存在于原核生物中,在胞壁酸合成中发挥重要的作用,Mtb中也称为转位酶Ⅰ(TranslocaseⅠ, MurX)。分枝杆菌属以异戊二烯基磷酸盐为原料,在MurX作用下,催化UDP-MurNGlyc-pentapeptide和UDP- MurNAc-pentapeptide合成相应的脂质Ⅰ[44-45]。脂质Ⅰ是处于第1位的膜锚定肽前体,在细胞壁合成中发挥重要的作用。该酶的作用受到抑制会直接影响菌体的生存。
该蛋白编码基因在Mtb中编号为Rv2156c,共1 080 bp,编码359个氨基酸。
三山霉素是我国贵州地区分离的链霉菌sp. SS的代谢产物,属于尿苷肽类抗生素,其可以通过抑制MurX的催化活性,发挥抗结核作用[46]。Shi等[46]通过基因工程方法,将链霉菌sp. SS中的基因缺失掉,获得了6种三山霉素的衍生物 (图4),其中MX-4和MX-6针对Mtb H37Rv的MIC为8 μg/mL,具有开发为新型抗结核药物的潜力。
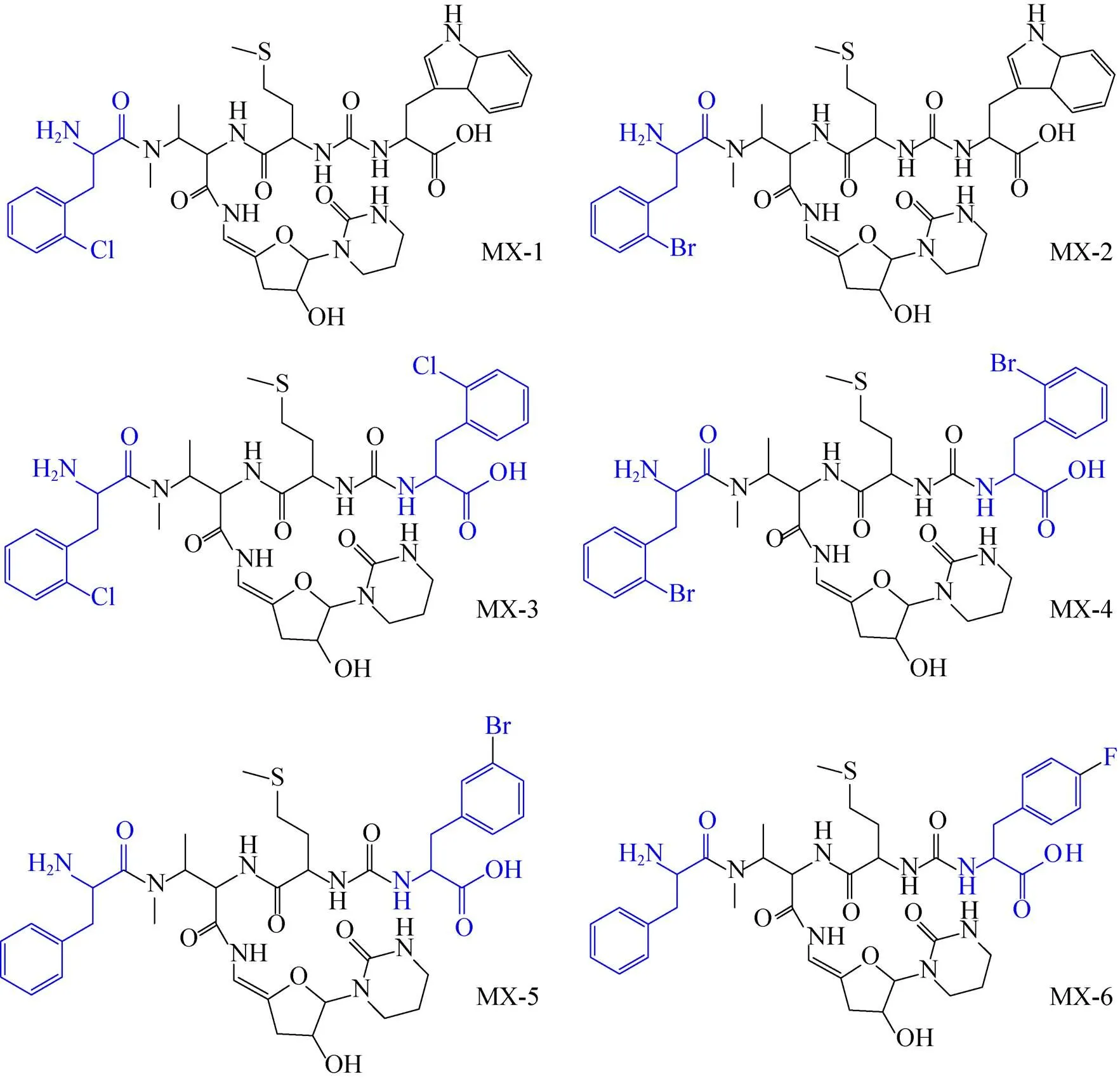
图4 尿苷肽类抗生素三山霉素
2.6 多异戊二烯基磷酸-GlcNAc-1-磷酸酯转移酶 (Polyprenyl phosphate-GlcNAc-1-phosphate transferase, WecA)
Mtb的细胞壁对于其在巨噬细胞内的生长是至关重要的[47-48],比较对数生长期和非复制期Mtb基因的表达情况发现,非复制期时与细胞壁合成和组装相关的基因 (糖转移酶、脂质阿拉伯甘露聚糖合成酶、分枝菌酸合成酶及其他细胞壁组装相关酶类) 表达明显上调[48-49]。因此,Mtb细胞壁合成中的关键酶类——分枝阿拉伯半乳聚糖合成酶的功能如果受到抑制,可以使处于非复制期的Mtb对现在所使用的抗结核药物变得敏感,从而杀死巨噬细胞内的Mtb。WecA是聚戊烯基磷酸酯N-乙酰己糖胺-1-磷酸酯转移酶,在Mtb中催化异戊二烯基-GlcNAc-焦磷酸酯 (Decaprenyl-GlcNAc-pyrophosphate, decaprenyl- P-P-GlcNAc) 的合成。WecA可以催化UDP- GlcNAc的Phospho-GlcNAc部分锚定到异戊二烯基磷酸盐 (Decaprenylphosphate) 上,从而起始分枝阿拉伯半乳聚糖的合成。之前的研究发现,该酶的功能受到抑制,会明显降低Mtb的生存能力[50]。因此,WecA是一个很好的药物靶标。该蛋白编码基因在Mtb中编号为Rv1302,共1 215 bp,编码404个氨基酸。
Mitachi等[51]构建了以WecA为靶标的高通量筛选模型,同时利用化学方法合成了抗结核核苷类抗生素Capuramycin的结构类似物UT-01320 (图5)。UT-01320在WecA筛选模型中表现出很强的抑制WecA酶的活性。在体外实验中,UT-01320在很低的浓度下,不仅能杀死处于复制期的Mtb (MIC=1.5 μg/mL),还可以杀死处于静止期的Mtb (MIC=2.58 μg/mL)。此外巨噬细胞内的Mtb也会被其消灭。其浓度为0.02 μg/mL时,针对Mtb的抑制率为42%;其浓度为2 μg/mL和20 μg/mL时抑制率可达到100%,其IC50为 (9.14±0.035 4) μg/mL。
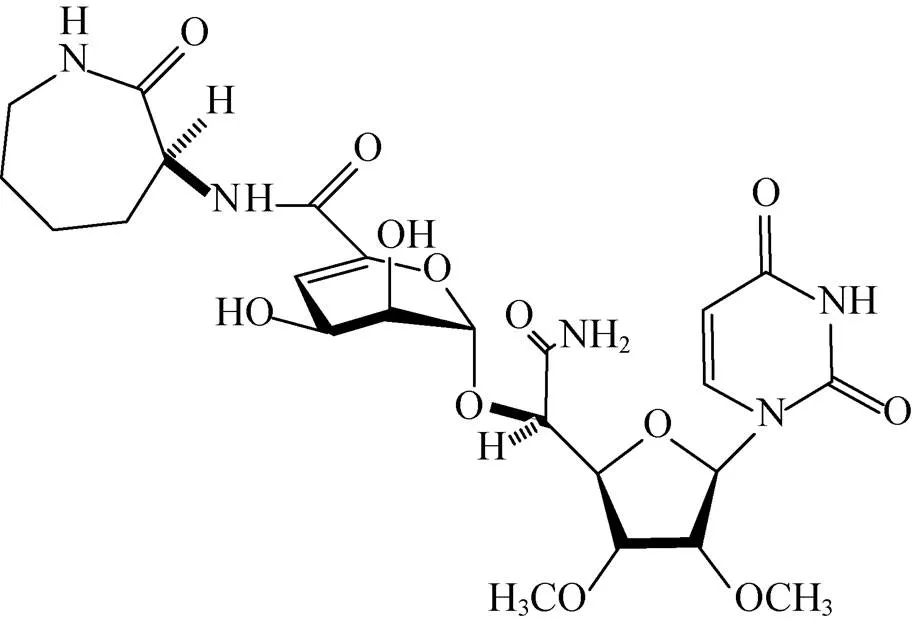
图5 UT-01320结构式
2.7 莽草酸激酶 (Shikimatekinase, AroK)
莽草酸激酶在ATP作为协同底物的情况下,可以将莽草酸转化为莽草酸-3-磷酸酯[52]。它属于核苷单磷酸 (Nucleoside monophosphate, NMP) 激酶家族成员,拥有3个发挥独立功能结构域:核心、顶盖和核苷单磷酸结合域[53]。核心区域由5条平行的β-折叠和1个核酸结合环 (P-loop) 构成,用于结合核苷酸;顶盖区域用于封盖活性中心,以调节与ATP的结合;核苷单磷酸结合区域则用于结合莽草酸[54-57]。
莽草酸途径在细菌、藻类、植物和寄生虫中普遍存在,而哺乳动物中缺乏该途径。该途径的最终代谢产物分枝酸是初级代谢产物预苯酸、邻氨基苯甲酸酯和氨基脱氧分枝酸的前体,而这3种初级代谢产物又可以用于进一步合成芳香氨基酸类、分枝菌素、泛醌、对氨苯甲酸、萘醌类等等与Mtb生长密切相关的化合物[58]。该途径的缺失会直接导致Mtb的死亡[59],故针对该途径的抑制剂可以作为潜在的抗结核药物进行开发。
该蛋白编码基因在Mtb中编号为Rv2539c,共531 bp,编码176个氨基酸。
Rajput等[60]以AroK为靶标构建了高通量的药物筛选模型,通过筛选容量为1 000个化合物的抗结核化合物库,筛选出两个极具潜力的化合物5631296 (图6) 和5491210 (图7)。5631296针对Mtb H37Rv的MIC=4 μg/mL,MBC=4 μg/mL,IC50为 (5.10±0.6) μmol/L,而5491210针对Mtb H37Rv的MIC=8 μg/mL,MBC=32 μg/mL,IC50为 (8.58±0.9) μmol/L,而对于MDR来讲,其MIC= 1 μg/mL。这对治疗MDR是一个十分振奋人心的发现。
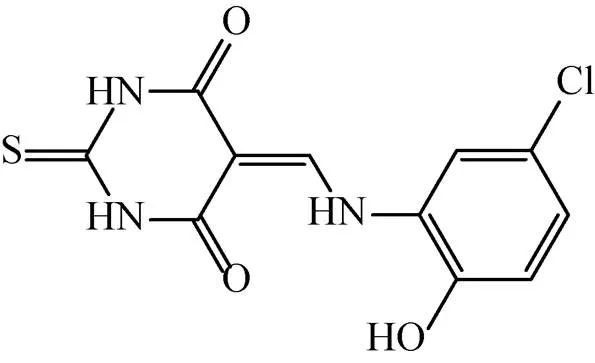
图6 化合物5631296结构式
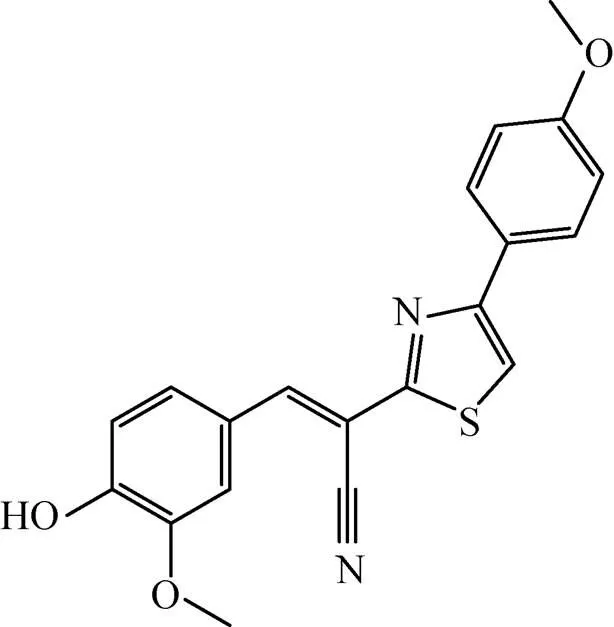
图7 化合物5491210结构式
3 展望
结核病虽然是一种可以防控和治疗的疾病,但是随着耐药性结核菌的大量出现以及结核病与其他疾病的合并感染,目前仍然没有得到很好的控制,还有进一步蔓延的趋势,形势依然严峻。世界卫生组织、世界各国的政府和研究机构对结核病研究的大量投入,为抗结核新药的开发带来了新的希望。新的抗生素靶标及其抑制剂不断涌现,除了前文提到的有明确作用位点的抑制剂外,还有一些能明显抑制Mtb生长,但作用位点未知的化合物,比如呋咱氮氧化物 (Furoxan) 衍生物[61]和两性分子氧杂蒽酮 (Xanthones)[62]。这一类作用机制或作用位点未知的化合物,依然有开发为抗结核药物的潜力,随着这类化合物作用机理的进一步研究,相信会涌现出更多的抗生素新靶标。原本用于治疗精神类疾病所用的药物甲硫哒嗪 (Thioridazine)[63],发现其也有抗结核的作用,这也为抗结核药物的开发提供了新的思路。随着蛋白质组学、功能基因组学在Mtb致病、耐药和持留方面研究中的广泛应用,相信定会在寻找高效靶点、建立高效药物筛选平台方面取得更大的突破。
[1] Global tuberculosis report 2017 [EB/OL]. [2017-12-01]. http://www.who.int/tb/publications/global_report/en/.
[2] Zhang Q, Wu ZY, Zhang ZR, et al. Efficacy and effect of free treatment on multidrug-resistant tuberculosis. Exp Ther Med, 2016, 11(3): 777–782.
[3] Gualano G, Capone S, Matteelli A, et al. New antituberculosis drugs: from clinical trial to programmatic use. Infect Dis Rep, 2016, 8(2): 6569.
[4] Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology offrom the complete genome sequence. Nature, 1998, 393(6685): 537–544.
[5] Flärdh K. Essential role of DivIVA in polar growth and morphogenesis inA3(2). Mol Microbiol, 2003, 49(6): 1523–1536.
[6] Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol, 2003, 48(1): 77–84.
[7] Kang CM, Nyayapathy S, Lee JY, et al. Wag31, a homologue of the cell division protein DivIVA, regulates growth, morphology and polar cell wall synthesis in mycobacteria. Microbiology, 2008, 154(3): 725–735.
[8] Singh V, Dhar N, Pató J, et al. Identification of aminopyrimidine-sulfonamides as potent modulators of Wag31-mediated cell elongation in mycobacteria. Mol Microbiol, 2017, 103(1): 13–25.
[9] Van Baarle S, Celik IN, Kaval KG, et al. Protein-protein interaction domains ofDivIVA. J Bacteriol, 2013, 195(5): 1012–1021.
[10] Jani C, Eoh H, Lee JJ, et al. Regulation of polar peptidoglycan biosynthesis by Wag31 phosphorylation in mycobacteria. BMC Microbiol, 2010, 10: 327.
[11] Sala C, Dhar N, Hartkoorn RC, et al. Simple model for testing drugs against nonreplicating. Antimicrob Agents Chemother, 2010, 54(10): 4150–4158.
[12] Olsen LR, Vetting MW, Roderick SL. Structure of thebifunctional GlmU acetyltransferase active site with substrates and products. Protein Sci, 2007, 16(6): 1230–1235.
[13] Olsen LR, Roderick SL. Structure of theGlmU pyrophosphorylase and acetyltransferase active sites. Biochemistry, 2001, 40(7): 1913–1921.
[14] Barreteau H, Kovač A, Boniface A, et al. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol Rev, 2008, 32(2): 168–207.
[15] Zhang WL, Jones VC, Scherman MS, et al. Expression, essentiality, and a microtiter plate assay for mycobacterial GlmU, the bifunctional glucosamine-1- phosphate acetyltransferase and-acetylglucosamine-1- phosphate uridyltransferase. Int J Biochem Cell Biol, 2008, 40(11): 2560–2571.
[16] Mehra R, Rani C, Mahajan P, et al. Computationally guided identification of novelGlmU inhibitory leads, their optimization, andvalidation. ACS Comb Sci, 2016, 18(2): 100–116.
[17] Domenech P, Reed MB, Barry III CE. Contribution of theMmpL protein family to virulence and drug resistance. Infect Immun, 2005, 73(6): 3492–3501.
[18] Lamichhane G, Tyagi S, Bishai WR. Designer arrays for defined mutant analysis to detect genes essential for survival ofin mouse lungs. Infect Immun, 2005, 73(4): 2533–2540.
[19] Varela C, Rittmann D, Singh A, et al.genes are associated with mycolic acid metabolism in mycobacteria and corynebacteria. Chem Biol, 2012, 19(4): 498–506.
[20] Tullius MV, Harmston CA, Owens CP, et al. Discovery and characterization of a unique mycobacterial heme acquisition system. Proc Natl Acad Sci USA, 2011, 108(12): 5051–5056.
[21] Owens CP, Chim N, Graves AB, et al. Thesecreted protein Rv0203 transfers heme to membrane proteins MmpL3 and MmpL11. J Biol Chem, 2013, 288(30): 21714–21728.
[22] Deidda D, Lampis G, Fioravanti R, et al. Bactericidal activities of the pyrrole derivative BM212 against multidrug-resistant and intramacrophagicstrains. Antimicrob Agents Chemother, 1998, 42(11): 3035–3037.
[23] La Rosa V, Poce G, Canseco JO, et al. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob Agents Chemother, 2012, 56(1): 324–331.
[24] Ramesh R, Shingare RD, Kumar V, et al. Repurposing of a drug scaffold: Identification of novel sila analogues of rimonabant as potent antitubercular agents. Eur J Med Chem, 2016, 122: 723–730.
[25] Hunter T. The Croonian Lecture 1997. The phosphorylation of proteins on tyrosine: its role in cell growth and disease. Philos Trans Roy Soc B Biol Sci, 1998, 353(1368): 583–605.
[26] Beresford NJ, Mulhearn D, Szczepankiewicz B, et al. Inhibition of MptpB phosphatase fromimpairs mycobacterial survival in macrophages. J Antimicrob Chemother, 2009, 63(5): 928–936.
[27] He RJ, Yu ZH, Zhang RY, et al. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol Sin, 2014, 35(10): 1227–1246.
[28] Singh R, Rao V, Shakila H, et al. Disruption ofimpairs the ability ofto survive in guinea pigs. Mol Microbiol, 2003, 50(3): 751–762.
[29] Singh R, Singh A, Tyagi AK. Deciphering the genes involved in pathogenesis of. Tuberculosis, 2005, 85(5/6): 325–335.
[30] Koul A, Herget T, Klebl B, et al. Interplay between mycobacteria and host signalling pathways. Nat Rev Microbiol, 2004, 2(3): 189–202.
[31] Zhou B, He YT, Zhang X, et al. Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc Natl Acad Sci USA, 2010, 107(10): 4573–4578.
[32] Castandet J, Prost JF, Peyron P, et al. Tyrosine phosphatase MptpA ofinhibits phagocytosis and increases actin polymerization in macrophages. Res Microbiol, 2005, 156(10): 1005–1013.
[33] Chauhan P, Reddy PV, Singh R, et al. Secretory phosphatases deficient mutant ofimparts protection at the primary site of infection in guinea pigs. PLoS ONE, 2013, 8(10): e77930.
[34] Bach H, Papavinasasundaram KG, Wong D, et al.virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe, 2008, 3(5): 316–322.
[35] Wong D, Bach H, Sun J, et al.protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci USA, 2011, 108(48): 19371–19376.
[36] Cowley SC, Babakaiff R, Av-Gay Y. Expression and localization of theprotein tyrosine phosphatase PtpA. Res Microbiol, 2002, 153(4): 233–241.
[37] Chen DN, Chen H, She ZG, et al. Identification of bostrycin derivatives as potential inhibitors ofprotein tyrosine phosphatase (MptpB). Med Chem, 2016, 12(3): 296–302.
[38] Auger G, van Heijenoort J, Mengin-Lecreulx D, et al. A MurG assay which utilises a synthetic analogue of lipid I. FEMS Microbiol Lett, 2003, 219(1): 115–119.
[39] van Heijenoort J. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol Mol Biol Rev, 2007, 71(4): 620–635.
[40] Bupp K, van Heijenoort J. The final step of peptidoglycan subunit assembly inoccurs in the cytoplasm. J Bacteriol, 1993, 175(6): 1841–1843.
[41] Raymond JB, Mahapatra S, Crick DC, et al. Identification of thegene, encoding the hydroxylase responsible for the-glycolylation of the mycobacterial peptidoglycan. J Biol Chem, 2005, 280(1): 326–333.
[42] Mahapatra S, Scherman H, Brennan PJ, et al. N Glycolylation of the nucleotide precursors of peptidoglycan biosynthesis ofspp. is altered by drug treatment. J Bacteriol, 2005, 187(7): 2341–2347.
[43] Mahapatra S, Crick DC, Brennan PJ. Comparison of the UDP--acetylmuramate:L-alanine ligase enzymes fromand. J Bacteriol, 2000, 182(23): 6827–6830.
[44] Kurosu M, Mahapatra S, Narayanasamy P, et al. Chemoenzymatic synthesis of Park’s nucleotide: toward the development of high-throughput screening for MraY inhibitors. Tetrahedron Lett, 2007, 48(5): 799–803.
[45] Bugg TDH, Lloyd AJ, Roper DI. Phospho-MurNAc- pentapeptide translocase (MraY) as a target for antibacterial agents and antibacterial proteins. Infect Disord Drug Targets, 2006, 6(2): 85–106.
[46] Shi YY, Jiang ZB, Lei X, et al. Improving the N-terminal diversity of sansanmycin through mutasynthesis. Microb Cell Fact, 2016, 15: 77.
[47] Grzegorzewicz AE, Ma YF, Jones V, et al. Development of a microtitre plate-based assay for lipid-linked glycosyltransferase products using the mycobacterial cell wall rhamnosyltransferase WbbL. Microbiology, 2008, 154(12): 3724–3730.
[48] Jin Y, Xin Y, Zhang WL, et al.Rv1302 andMSMEG_4947 have WecA function and MSMEG_4947 is required for the growth of. FEMS Microbiol Lett, 2010, 310(1): 54–61.
[49] Bacon J, Alderwick LJ, Allnutt JA, et al. Non-replicatingelicits a reduced infectivity profile with corresponding modifications to the cell wall and extracellular matrix. PLoS ONE, 2014, 9(2): e87329.
[50] Ishizaki Y, Hayashi C, Inoue K, et al. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J Biol Chem, 2013, 288(42): 30309–30319.
[51] Mitachi K, Siricilla S, Yang D, et al. Fluorescence-based assay for polyprenyl phosphate-GlcNAc-1-phosphate transferase (WecA) and identification of novel antimycobacterial WecA inhibitors. Anal Biochem, 2016, 512: 78–90.
[52] Pereira JH, Vasconcelos IB, Oliveira JS, et al. Shikimate kinase: a potential target for development of novel antitubercular agents. Curr Drug Targets, 2007, 8(3): 459–468.
[53] Vonrhein C, Schlauderer GJ, Schulz GE. Movie of the structural changes during a catalytic cycle of nucleoside monophosphate kinases. Structure, 1995, 3(5): 483–490.
[54] Pereira JH, De Oliveira JS, Canduri F, et al. Structure of shikimate kinase fromreveals the binding of shikimic acid. Acta Crystallogr D Biol Crystallogr, 2004, 60(12): 2310–2319.
[55] Hartmann MD, Bourenkov GP, Oberschall A, et al. Mechanism of phosphoryl transfer catalyzed by shikimate kinase from. J Mol Biol, 2006, 364(3): 411–423.
[56] Dhaliwal B, Nichols CE, Ren JS, et al. Crystallographic studies of shikimate binding and induced conformational changes inshikimate kinase. FEBS Lett, 2004, 574(1/3): 49–54.
[57] Gan JH, Gu YJ, Li Y, et al. Crystal structure ofshikimate kinase in complex with shikimic acid and an ATP analogue. Biochemistry, 2006, 45(28): 8539–8545.
[58] Kapnick SM, Zhang Y. New tuberculosis drug development: targeting the shikimate pathway. Expert Opin Drug Discov, 2008, 3(5): 565–577.
[59] Parish T, Stoker NG. The common aromatic amino acid biosynthesis pathway is essential in. Microbiology, 2002, 148(10): 3069–3077.
[60] Rajput VS, Mehra R, Kumar S, et al. Screening of antitubercular compound library identifies novel shikimate kinase inhibitors of. Appl Microbiol Biotechnol, 2016, 100(12): 5415–5426.
[61] dos Santos Fernandes GF, de Souza PC, Marino LB, et al. Synthesis and biological activity of furoxan derivatives against. Eur J Med Chem, 2016, 123: 523–531.
[62] Koh JJ, Zou HX, Mukherjee D, et al. Amphiphilic xanthones as a potent chemical entity of anti- mycobacterial agents with membrane-targeting properties. Eur J Med Chem, 2016, 123: 684–703.
[63] de Keijzer J, Mulder A, de Haas PEW, et al. Thioridazine alters the cell-envelope permeability of. J Proteome Res, 2016, 15(6): 1776–1786.
(本文责编 陈宏宇)
Progress in role of novel anti-tuberculosis drug targets and their inhibitors
Lei Ji1, and Jianping Xie2
1 Department of Basic Medicine, Hangzhou Medical College, Hangzhou 310053, Zhejiang, China 2 State Key Laboratory Breeding Base of Eco-Environment and Bio-Resource of Three-Gorges Area, Institute of Modern Biopharmaceuticals, School of Life Sciences, Southwest University, Chongqing 400715, China
There is an urgent need to discover new anti-tuberculosis compounds with novel mechanisms of action in order to deal with tuberculosis and drug-resistant tuberculosis. In this article, the feature of several promising novel compounds with potential anti-tuberculosis activities is summarized, focusing on the drug targets of these compounds. These summaries will contribute to the further development of anti-tuberculosis drugs.
inhibitors, drug target, tuberculosis
December 27, 2017;
March 26, 2018
National Natural Science Foundation of China (Nos. 81701970, 81371851), Scientific Research Foundation of the Education Department of Zhejiang Province (No. Y201534356).
Jianping Xie. Tel/Fax: +86-23-68367108; E-mail: georgex@swu.edu.cn
国家自然科学基金 (Nos. 81701970, 81371851),浙江省教育厅一般科研项目 (No. Y201534356) 资助。
10.13345/j.cjb.170521
猜你喜欢
当代水产(2022年1期)2022-04-26 14:35:38
黑龙江大学自然科学学报(2021年4期)2021-11-19 07:05:02
昆明医科大学学报(2021年4期)2021-07-23 01:22:00
军民两用技术与产品(2021年10期)2021-03-16 06:05:10
世界农药(2019年3期)2019-09-10 07:04:10
中国调味品(2017年2期)2017-03-20 16:18:21
现代检验医学杂志(2016年3期)2016-11-15 01:59:48
光学精密工程(2016年4期)2016-11-07 09:04:48
中国民族医药杂志(2016年4期)2016-05-09 07:41:11
中国医药生物技术(2015年4期)2015-12-26 08:26:36
