
磷钨酸修饰铁改性类石墨相氮化碳的制备、表征及光催化性能研究
2018-08-23 12:10蔡天凤李会鹏
精细石油化工 2018年4期
张 杰,蔡天凤,李会鹏, 赵 华
(辽宁石油化工大学化学化工与环境学部,辽宁 抚顺 113001)
1972年Fujishima等[1]发表了关于TiO2能够光催化电解水的研究,从此半导体的光催化应用便进入人们的视野。类石墨相氮化碳(g-C3N4)是一种无污染不含金属的绿色催化剂。1996年,Teter和Hemley[2]对氮化碳的结构进行了严密的计算,发现C3N4存在5种同素异形体,分别为:α-C3N4、β-C3N4、g-C3N4、cubic-C3N4(立方相)和 pseudo-cubic-C3N4(准立方相),提出g-C3N4是最稳定的结构,从此科学家将目光集中到g-C3N4结构的应用与表征上。g-C3N4表现为二维层状结构,在分子内部层间以N为杂原子进行共轭π键连接,在碳原子与氮原子之间由共价键进行连接[3],在分子与分子之间存在弱的相互作用[4]。由于其组织结构以及电子结构的特殊性,使其即使在酸碱条件下也能保持高的稳定性。目前,g-C3N4在光催化水解制氢制氧[5-7]、光催化污染物的降解[8]、光催化有机合成等[9]领域都有广泛的应用。但是g-C3N4也同样存在一定的缺陷,例如具有较低的有效比表面积;光生电荷极易复合;只能吸收蓝紫光,极大的降低了光能的利用率[10-13]。
杂多酸是一种绿色新型环保催化剂,由杂原子和多原子通过氧原子桥联配位的一类含氧多原子酸。具有配合物和金属氧化物的特征,表现出特有的物理以及化学活性,在催化氧化领域受到青睐[14]。但是杂多酸又有其局限性,比如杂多酸比表面积较小,易溶于水、丙酮等极性较强的小分子溶剂中并且不宜分离。所以杂多酸的固化,有效解决了杂多酸自身的问题。通过将杂多酸固载在多孔载体上,可大大提高其比表面积,使其具有更高的催化活性以及选择性。Zhang等[15]将磷钨酸固载在ZrO2修饰的SiO2上,制得了H3PW12O40/ZrO2-SiO2催化剂进行氧化脱硫,在最佳条件下,二苯并噻吩的脱除率达到100%。
针对g-C3N4以及杂多酸优点和缺点,本实验以g-C3N4为载体,通过浸渍法负载Fe在载体上,增大比表面积,降低光生电子-空穴的复合几率,提高量子效率。再以g-C3N4表面含有大量氨基的特点[16],将磷钨酸(HPW)引入到Fe-C3N4中制备HPW/Fe-C3N4催化剂,依靠HPW本身具有的氧化性及酸性,增加对反应底物的催化氧化性能同时与g-C3N4半导体协同催化。采用了紫外可见(UV-Vis)漫反射光谱、X射线衍射(XRD)光谱、红外傅里叶变换(FTIR)光谱、荧光(PL)光谱、扫描电镜(SEM)进行表征,并且以罗丹明B为探针对催化剂的反应活性进行了考察。
1 实验部分
1.1 主要试剂
三聚氰胺(C3H6N6),上海试剂一厂;磷钨酸(H3PO4·12WO3·xH2O)、硝酸铁(Fe(NO3)3·9H2O),天津市大茂化学试剂厂;无水乙醇(CH3CH2OH),中国医药公司。
1.2 仪器及表征
X射线衍射光谱(XRD)采用德国布鲁克公司的D8 Advance进行测定,以Cu靶作为辐射电源;傅里叶变换红外(FT-IR)使用美国尼高力的TP-FTIR傅里叶红外光谱仪进行测定,采用ATR附件;紫外可见(UV-Vis)漫反射光谱美国安捷伦公司的Agilent Cary 5000进行测定;荧光(PL)光谱采用的是安捷伦科技有限公司(美国)Cary Eclipse的荧光分光光度计进行测定,氙灯为激发光源;扫描电镜(SEM)采用日本日立公司的发射扫描电镜SU8010进行测定,观察粉体的表面形貌,加速电压为15 kV。
1.3 HPW/xFe-C3N4催化剂的制备
1)分别称取Fe(NO3)3·9H2O(质量分别为0.374 2、0.778 5、1.213 6、1.681 0 g)溶解在30 mL无水乙醇中,加入10 g三聚氰胺,搅拌3 h直至乙醇蒸干,在60 ℃下烘干。在升温速率为2 ℃/min,550 ℃下煅烧2 h。得到的黄色粉末即为不同Fe负载量的x%Fe-C3N4(x=0、0.5%、1.0%、1.5%、2.0%)。
2)称取4g的x%Fe-C3N4溶解在一定量去离子水中。称取4 g的HPW在10 mL水和10 mL乙醇的混合溶液中充分溶解,倒入含有Fe-C3N4的溶液,超声1 h,并在室温下搅拌3 h直到水分完全蒸发, 60 ℃下干燥。将干燥的后固体在300 ℃下煅烧2 h,升温速率为5 ℃/min。待自然降至室温即得到目标产物。
1.4 催化剂的活性评价
降解率的计算:
η=(A0-A)/A0
式中,η为降解率,A0为原样的吸光度,A为降解后的吸光度
活性评价:以500 W的氙灯为光源,以RhB为探针,对催化剂进行活性评价。称取0.02 g的催化剂加入到25 mL浓度为10 mg/L的RhB溶液中,在暗光下搅拌30 min达到催化剂对反应底物的吸附平衡。之后在氙灯的照射下进行光催化反映,每30 min取样一次,用紫外可见分光光度计在550 nm的波长下测量其吸光度。
1.5 动力学分析
从动力学角度对催化剂进行活性评价,光催化降解符合一级反应动力学。应用Langmuir-Hinshelwood模型,对水相下光催化降解进行动力学分析
式中,r0是最初的反应速率,mg/(L·min);C是罗丹明B的浓度,mg/L;t是反应时间,min;k是Langmuir-Hinshelwood反应速率常数,mg/(L·min);K是Langmuir吸附平衡常数,L/mg。罗丹明B被稀释后把公式简化为:
式中,kapp为表观速率常数,min-1;C0是罗丹明B的初始浓度。
2 结果与讨论
2.1 催化剂g-C3N4 和 HPW/x%Fe-C3N4 的表征
2.1.1FT-IR
图1为g-C3N4和HPW/x%Fe-C3N4的FT-IR图谱。在g-C3N4的红外光谱中,吸收特征峰主要集中在808、1 240~1 650、3 170 cm-1区域,其中3 170 cm-1附近的吸收特征峰对应的是未完全缩聚的氨基内的N—H的伸缩振动[17,18],是磷钨酸引入氮化碳结构的关键官能团。HPW/1.5%Fe-C3N4仍然在700~1 100 cm-1出现了Keggin结构的4个特征峰,并且能明显观察到795 cm-1处P—O—P键的反对称伸缩振动吸收特征峰[19],而HPW/0.5%Fe-C3N4、HPW/1.0%Fe-C3N4、HPW/2.0%Fe-C3N4的FT-IR谱图中没有观察到P—O—P键的反对称伸缩振动吸收特征峰(图B)。此外,HPW/1.5%Fe-C3N4在1 535 cm-1并没有出现氮化碳的吸收特征峰,表明HPW/1.5%Fe-C3N4氮化碳的结构已经发生了变化,但是却保留了HPW的4个吸收特征峰,即仍然保持磷钨酸的Keggin结构。
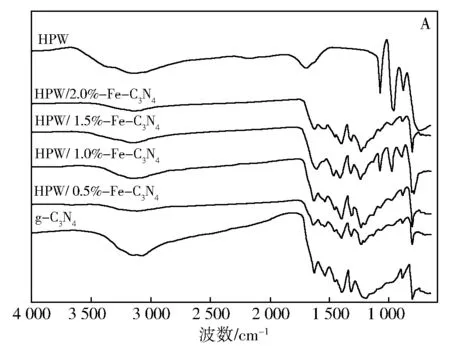
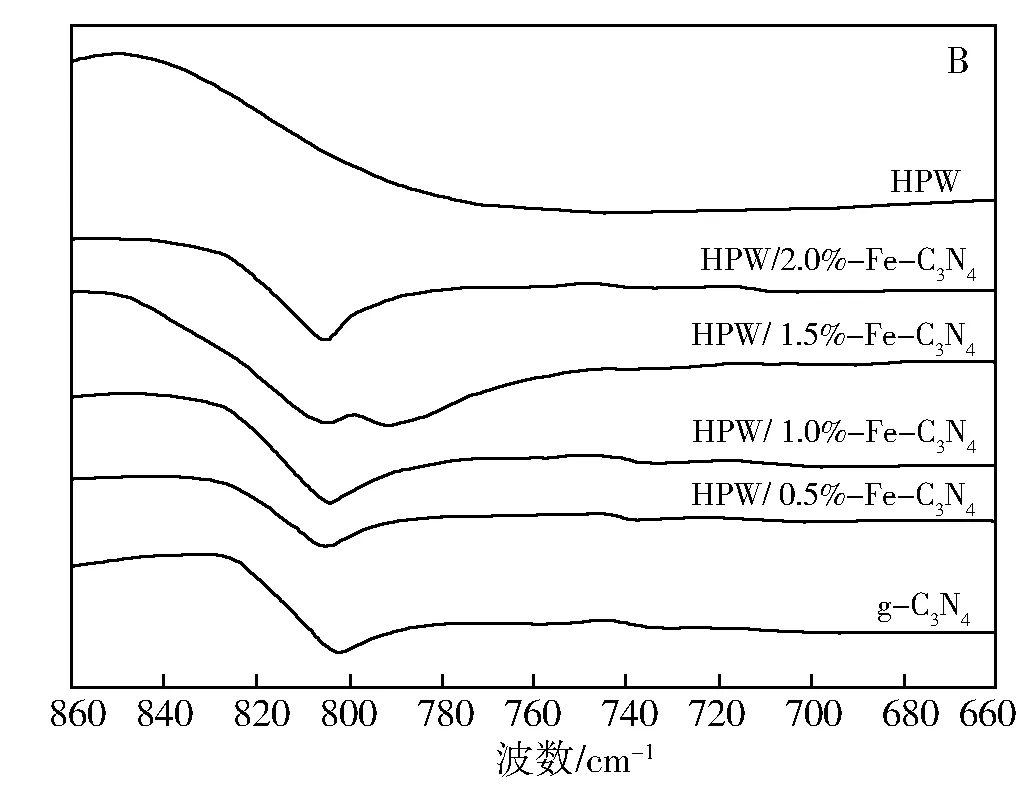
图1 g-C3N4和HPW/x3N4催化剂的FT-IR谱
2.1.2XRD
图2为g-C3N4基样品的XRD谱。在27.5°、13.2°分别出现了g-C3N4的特征衍射峰,其中13.2°附近的衍射峰对应晶面指数为100,是平面内三均三嗪结构之间的氮孔间距d=0.67 nm[20]。
相对于g-C3N4,HPW/x%Fe-C3N4在13.2°附近的衍射峰逐渐减弱直至消失,在27.5°附近衍射峰宽化,表明Fe的掺杂抑制了氮化碳晶粒的生长[21],扩大了层间距。并且在HPW/x%Fe-C3N4催化剂中并没有发现磷钨酸的特征峰,表明磷钨酸已经从晶体变为非定性态,并且以化学键的形式很好的负载在载体上[22]。HPW/1.5%Fe-C3N4在7°~12°、16°~22°、25°~30°处具有特征衍射峰,与Keggin型结构的特征衍射峰一致,表明HPW、Fe、g-C3N4形成了具有Keggin结构的杂多酸盐,但是g-C3N4的特征峰很弱,所以可以认为在形成盐的同时,g-C3N4的结构发生了变化,与FT-IR的结果一致。而在其他的催化剂中并没有观察到完整的Keggin型结构的特征衍射峰,这主要是因为当x<1.5时,杂多酸盐的含量较少且均匀的分散在g-C3N4表面,峰位被g-C3N4的峰覆盖,而当x>1.5时,大量的磷钨酸与Fe反应,而未能与g-C3N4上的氨基反应。
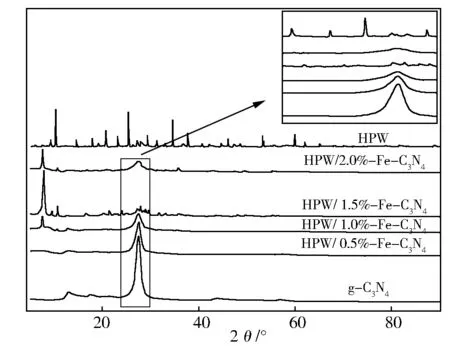
图2 g-C3N4和HPW/x%Fe-C3N4催化剂的XRD谱
2.1.3UV-Vis
图3为催化剂UV-Vis图谱,右上为不同Fe含量的催化剂样品。由图3可以看出,随着Fe含量的增加,样品的颜色也逐渐加深。在UV-Vis谱图中,样品的吸光带并没有表现出明显的拐点,表明Fe、g-C3N4、HPW三者之间是以化学键的形式链接在一起而非简单的机械混合[23]。相对于g-C3N4,HPW/x%Fe-C3N4表现出了明显的红移现象,吸收阈值从460 nm左右拓宽到500 nm,在300~500 nm表现出强烈的吸收性能,表明Fe以及磷钨酸的掺杂可以增加石墨相氮化碳的对于光的敏感性,提高对光的捕获能力。利用截线法测量材料的吸收阈值,各个材料的吸收阈值以及带隙能如表1。
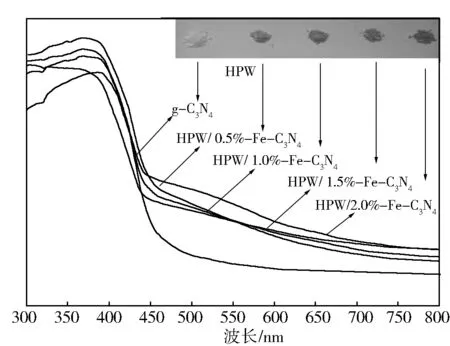
图3 g-C3N4和HPW/x%Fe-C3N4催化剂的 UV-Vis漫反射谱
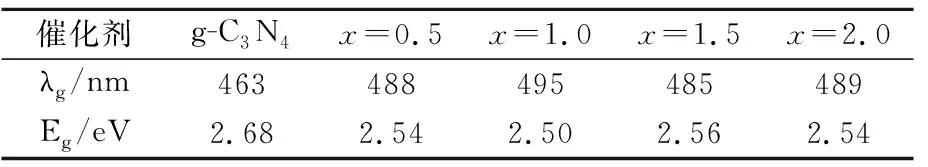
催化剂g-C3N4x=0.5x=1.0x=1.5x=2.0λg/nm463488495485489Eg/eV2.682.542.502.562.54
从表1可以看出,g-C3N4的禁带宽度为2.68 eV,与文献报道2.7eV基本一致[24, 25],并且可知Fe和HPW的掺杂改变了催化剂的能带结构,降低了带隙能,提高了可见光的利用率。
2.1.4PL
图4为g-C3N4以及HPW/x%Fe-C3N4的PL荧光发射光谱,激发光源波长为360 nm。g-C3N4在460 nm处具于很强的荧光发射峰,与UV-Vis谱图中g-C3N4的吸收阈值λg=463 nm的结果一致。HPW/x%Fe-C3N4催化剂相对于g-C3N4表现出略微的蓝移现象,并且荧光发射峰明显降低,主要是因为导带与价带之间出现了相对的位移,减小了禁带宽度,降低了带隙能而出现了量子限制效应[26],表现的结果为HPW/x%Fe-C3N4催化剂出现了明显的淬灭现象。表明Fe离子以及磷钨酸的掺杂有效捕获了光生载流子,抑制了光生电子-空穴的复合。
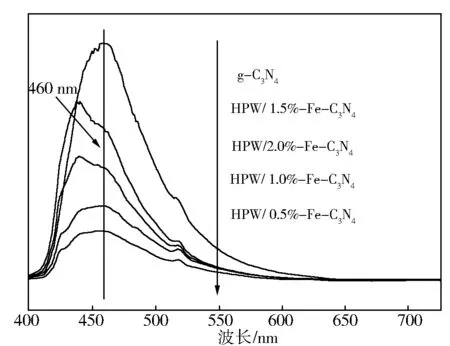
图4 g-C3N4和HPW/x%Fe-C3N4催化剂的PL发射图谱
2.1.5SEM
通过SEM扫描电镜对g-C3N4和HPW/x%Fe-C3N4的形貌进行考察,结果如图5所示。图5A为g-C3N4的SEM图,表现出不规则的块状结构。图5B、C、D、E是不同Fe含量的HPW/x%Fe-C3N4催化剂,相对于块状的g-C3N4表现出更松散稀疏的状态,原因可能在于一方面是超声的结果[27],另一方面是由于Fe的掺杂以及磷钨酸盐的形成。
2.2 g-C3N4和HPW/x%Fe-C3N4的催化活性评价
g-C3N4和HPW/x%Fe-C3N4系列催化剂在500 W氙灯照射下对罗丹明B的降解反应结果如图6所示。从图6可以看出,在不添加任何催化剂的情况下,罗丹明B几乎没有表现出降解。图6(A)中在光照之前,罗丹明B的浓度降低,主要是在催化剂表面发生了吸附作用。罗丹明B在g-C3N4,HPW/0.5%Fe-C3N4,HPW/1.0%Fe-C3N4,HPW/1.5%Fe-C3N4,HPW/2.0%Fe-C3N4表面的吸附率分别为1.75%、50.92%、52.18%、66.11%、52.77%。HPW/x%Fe-C3N4系列催化剂的吸附能力明显大于g-C3N4。经过2h的照射,降解率分别为:HPW/1.5%Fe-C3N4(96.10%)>HPW/2.0%Fe-C3N4(82.74%)> HPW/1.0%Fe-C3N4(74.19%)> HPW/0.5%Fe-C3N4(60.3%)>g-C3N4(58.72%)

图5 g-C3N4(A)和HPW/x%Fe-C3N4的SEM照片x=0.5%(B)、x=1.0%(C)、x=1.5%(D)、x=2.0%(E)
当x在0.5~1.5时,随着负载量的增加,光催化活性逐渐增加,但是当x=2.0时,催化活性反而降低,可能的原因在于一方面Fe的高掺杂量堵塞了孔道,另一方面在于Fe的含量过高,而形成了电子-空穴的复合中心。最佳的铁的负载量为1.5%,在2 h的照射下降解率达到了96.10%。原因在于,Fe和HPW的掺杂,增加了对反应底物的吸附能力,抑制了光生电子—空穴的复合[28],并且磷钨酸是具有强氧化性与酸性的半导体材料,与g-C3N4半导体发生协同作用[29]。
应用一级反应动力学对催化剂进行动力学活性评价,结果如图6(B)所示。g-C3N4,HPW/0.5%Fe-C3N4,HPW/1.0%Fe-C3N4,HPW/1.5%Fe-C3N4,HPW/2.0%Fe-C3N4对应的一级反应速率常数分别为0.005 5、0.017 8、0.020 7、0.025 6,0.039 4 min-1, HPW/x%Fe-C3N4催化剂的反应速率常数明显大于g-C3N4的速率常数,HPW/1.5%Fe-C3N4表现出最大反应速率,是g-C3N4的7倍,远远大于文献中记载的3.2倍[17]。

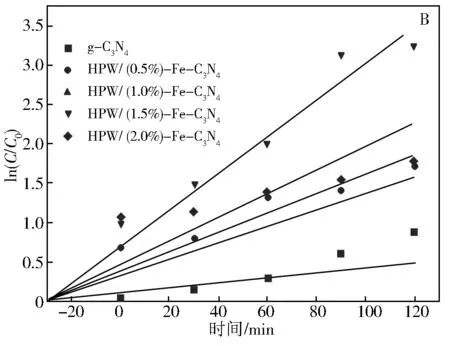
图6 g-C3N4和HPW/x%Fe-C3N4光照下的 催化活性评价以及降解RhB的一级反应动力学曲线
4 结 论
以三聚氰胺、Fe(NO3)3·9H2O 和HPW为原料,通过浸渍法制备了不同Fe含量的HPW/x%Fe-C3N4催化剂。结果表明,Fe以及HPW的掺杂,大大降低了带隙能,降低了光生电子-空穴复合的几率,吸收波长发生红移,提高了对于光能的利用率。并且当x=1.5时,由于磷钨酸盐与g-C3N4出现协同催化作用,使得光催化剂的活性进一步提高,降解率在2 h的照射下达到96.10%,速率常数为0.039 4 min-1,是纯g-C3N4的7倍。
猜你喜欢
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
安徽化工(2022年2期)2022-04-12
石油化工高等学校学报(2021年3期)2021-07-15
陶瓷学报(2021年1期)2021-04-13
粉末冶金技术(2021年1期)2021-03-29
世界核地质科学(2019年4期)2019-12-19
热处理技术与装备(2019年1期)2019-03-14
沈阳大学学报(自然科学版)(2018年5期)2018-11-07
电子制作(2018年12期)2018-08-01
