
僵人综合症1例报道
2018-08-02 03:12:48柯高潭吴茜陈博卜碧涛
神经损伤与功能重建 2018年7期
柯高潭,吴茜,陈博,卜碧涛
作者单位华中科技大学同济医学院附属同济医院神经内科武汉430030
僵人综合征(stiff-person syndrome,SPS)是一种以躯体中轴部肌肉进行性、波动性僵硬伴阵发性痉挛为特征的中枢神经系统少见疾病;突然的刺激可引起痛性痉挛发作,而睡眠时症状可消失。早期表现缺乏特异性,易出现误诊。现将我院诊治的1例SPS患者报道如下。
1 临床资料
患者,女,23岁,因“腰部肌肉僵硬1年”入院。1年前无明显诱因出现腰背部阵发性疼痛,卧床休息不能缓解。3 d后出现腰背部肌肉僵硬,同时弯腰、下蹲、转身、迈步等动作困难,无四肢麻木无力,夜间睡眠上述症状稍缓解,至我院风湿科就诊,行骶髂关节MRI提示左骶髂关节面骨质水肿,考虑脊柱关节病,给予洛索洛芬钠片、注射用重组人Ⅱ型肿瘤坏死因子受体抗体融合蛋白、柳氮磺吡啶治疗,腰部肌肉僵硬仍逐渐加重,并出现翻身困难,不能独自行走。既往史:身体健康,无外伤史,无家族史。体格检查:心肺及颅神经体征正常,四肢肌力正常;双侧腰部、臀部、腹部肌肉木板样紧张,触诊僵硬,左侧为主;双下肢肌张力呈铅管样增高,左侧为主(图1A);双上肢肌张力正常;四肢腱反射亢进,双侧病理征阴性;深浅感觉正常,共济失调阴性,颈软;脊柱侧弯及过度前凸,腰椎活动受限。辅助检查:血常规、肝肾功能、电解质、尿常规、粪常规、输血全套均正常。肿瘤全套示糖链抗原19-9 42.79 U/mL(正常值2.0~37.0 U/mL)。甲状腺功能正常,甲状腺免疫示甲状腺球蛋白抗体280.50 IU/mL(正常值10~115 IU/mL),甲状腺过氧化物酶抗体>600 IU/mL(正常值5~34 IU/mL)。风湿全套、ANCA、抗心磷脂抗体、类风湿全套、血沉均阴性。HLA-B27阴性。血清铜、血清铁、铁蛋白均正常。血抗谷氨酸脱羧酶(glutamic acid decarboxylase,GAD)抗体阳性。心电图、胸片正常。乳腺彩超示双侧乳腺增生。腹部彩超、子宫及附件彩超均未见明显异常。骶髂关节MRI示双侧骶髂关节面骨质硬化。双肾MRI未见明显异常。头部MRI平扫+增强未见明显异常。肌电图示双侧胫、腓神经运动感觉神经传导正常;双侧腰骶脊旁肌静息时可见不自主复合电位。治疗与随访:入院后给予巴氯芬5 mg 3次/天,奥沙西泮15 mg 2次/天治疗,第3天可翻身并独自缓慢行走,查体肌肉僵硬较前缓解。后给予血浆二重滤过3次,过程为每次处理血浆2 500 mL,丢弃血浆400 mL,补充人血白蛋白100 mL;同时口服强的松30 mg 1次/天。1月后我院神经内科门诊复诊,症状明显改善,可弯腰、下蹲,双下肢可弯曲、上抬,行走自如(图1B)。查体腰背部肌肉轻度紧张,双下肢肌张力正常。复查GAD抗体阳性,加用他克莫司3 mg 1次/天,随访1年,未出现复发。
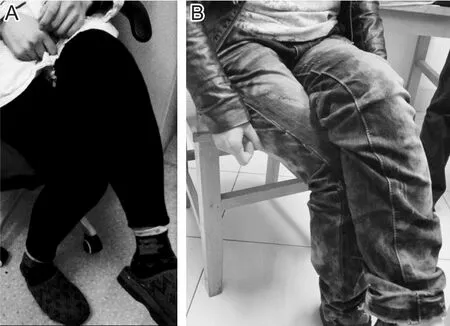
图1 患者肌张力检查
2 讨论
SPS是1956年由Moersch和Woltman命名[1]的一种自身免疫性疾病,临床少见,发病率约为1/100万[2]。其免疫机制尚不清楚,常常合并其它自身免疫性疾病,包括糖尿病、甲亢、甲减、恶性贫血、白癜风等。其中,约60%~80%SPS患者GAD抗体阳性[1],GAD是谷氨酸合成抑制性神经递质γ-氨基丁酸(gamma-aminobutyric acid,GABA)的限速酶;对其致病机制,部分学说[3]认为是由于突触囊泡分泌过程中GAD抗原的暴露,诱导机体产生GAD抗体,该抗体与GABA能神经元神经末梢的GAD发生免疫反应,导致GABA合成减少[4]及转运障碍[5],从而引起肌肉持续性兴奋。另外,SPS可以是副肿瘤综合征的一种表现,可出现在小细胞肺癌、乳腺癌、卵巢癌等患者中;且部分患者存在副肿瘤综合征相关抗体,如Gephyrin抗体和Amphiphysin抗体等,这些抗体通过影响GABA及甘氨酸等抑制性神经递质产生相应临床症状[6]。Feng等[7]研究对Gephyrin基因敲除的小鼠和对照组小鼠同时给予轻微的触觉刺激,结果显示Gephyrin基因敲除的小鼠表现为肢体伸直、僵硬,且出生12 h后上述反应更强烈,甚至出现呼吸困难;而对照组小鼠肢体未见明显反应。Geis等[8]研究对大鼠鞘内注射抗Amphiphysin的IgG抗体,可诱导大鼠产生肌肉强直痉挛症状,且躯干和后肢肌肉僵硬更明显。
SPS临床表现为以下几点:①中年女性多见,进行性轴性肌肉僵硬,突然的刺激引起阵发性痛性痉挛[6,9],部分患者僵硬首先累及肢体称为僵肢综合征(stiff limb syndrome,SLS);②可被临床观察及肌电图证实的主动肌与拮抗肌共同收缩[3];③其它神经系统疾病不能解释这些临床症状;④抗GAD抗体阳性[3];⑤苯二氮卓类药物治疗有效[9]。其中,SPS肌电图表现为静息时可持续出现正常运动单位电位,痉挛发作时运动单位发放明显增加,静注苯二氮卓类药物后运动单位明显减弱至停止。
SPS需要鉴别的疾病有进展型脑脊髓炎(progressive encephalomyelitis with rigidity and myoclonus,PERM)、运动神经元病、遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP)、锥体外系病变或帕金森病、Issac综合征、破伤风、遗传性惊跳病等[3,9,10]。①PERM具备SPS躯干僵直、痛性痉挛的特征,且肌电图结果同SPS,但有脑干症状、感觉症状、锥体束征、自主功能障碍、神经精神障碍、脑脊液细胞数增多等特征。②运动神经元病虽可出现肌肉僵硬,但以肌肉萎缩为主、且可出现球麻痹症状,常规肌电图提示在4个区域(脑干运动神经核、脊髓颈段、胸段或腰骶段的前角细胞)中至少2个区域出现神经源性损害。③椎体外系病变表现为肌张力增高甚至肌强直,但不出现痛性痉挛及肌肉波动性僵硬。④HSP是一组神经系统遗传性疾病,临床主要表现为双下肢痉挛,一般不累及躯干肌。⑤Issacs综合征临床表现为肌肉抽搐、痉挛和肌纤维颤搐,主要影响远端肌肉,睡眠时症状不消失,对苯妥英钠反应良好,有家族遗传史;肌电图表现为肌强直放电以及多联电位,持续的运动单位发放在远端比脊旁肌明显,运动及感觉传导速度减慢。⑥破伤风临床表现为全身肌肉痉挛强直,多有外伤病史,常出现咬肌痉挛症状,肌电图表现为H-反射消失,潜伏期缩短及运动终板机能异常(重复刺激可强化)。⑦遗传性惊跳病,主要以婴儿发病为主,以广泛性肌肉强直和夜间肌阵挛为特点的一种过度惊跳反射。⑧部分肌病,如强直脊柱综合征、纤维肌炎、Emery-Deifuss肌营养不良、少许炎性肌病等可出现躯干部肌肉强直、痉挛,这类肌肉收缩在睡眠时及麻醉情况下均存在,临床表现为肌肉萎缩,常规肌电图呈肌源性损害,实验室检查肌酶可升高,肌活检可明确诊断。
本例患者躯干及肢体肌肉僵硬,阵发性痛性痉挛,肌电图示静息时不自主复合电位活动,给予奥沙西泮治疗明显好转,无外伤史,头颅影像学正常,符合SPS的诊断。
僵人综合症的一线治疗是GABA受体激动剂,包括苯二氮卓类药物和巴氯芬,用于控制临床症状。对于病情严重的和难治性僵人综合症,丙种球蛋白是较好的二线治疗方案[1,11],血浆交换可作为丙种球蛋白的替代治疗,后期可加用免疫抑制剂[12,13]。激素常作为单药或联合其他治疗用于僵人综合症。临床可通过患者言语、平衡功能的改善及僵硬、痉挛次数的减少对疗效进行评价。
僵人综合症虽然罕见,但致残率和死亡率高,约10%的患者因持续痉挛或突然撤药引起突发死亡。所以,正确诊断、及时治疗对疾病预后具有重要作用。
