
基于PCR和位点特异性引物延伸反应的SNP检测方法的建立
2018-07-09 01:29:04单洪波金亚南
检验医学 2018年6期
单洪波, 金亚南
(1. 杭州艾迪康医学检验中心,浙江 杭州 310023;2. 杭州优思达生物技术有限公司,浙江 杭州 310053)
单核苷酸多态性(single nucleotide polymorphism,SNP)是指在基因组水平上,特定核苷酸位置上存在2种或2种以上不同的碱基,且其中任何一种等位基因在群体中的频率不<1%。SNP是一种单碱基水平的分子遗传标记,不仅可通过连锁或关联分析来定位疾病易感基因,而且有些SNP本身就可能会导致某些疾病或引起个体对药物反应的差异[1]。同时,SNP图谱的建立还能有效地帮助破解人类生理学密码,了解人类进化的起源以及对患者进行有效的治疗[2]。因此,SNP检测对疾病的风险性评估、诊断、预防和治疗等各方面均有很大的价值。
目前已报道了多种检测SNP的方法用于基因检测和药物基因组学的研究[1]。引物特异性聚合酶链反应(allele-specific polymerase chain reaction,AS-PCR)是目前比较常见的一种SNP检测技术,具体指通过设计针对不同SNP位点基因型的特异性引物达到特异性扩增和检测的目的[3-4]。改进的AS-PCR也包括引物扩增受阻突变体系聚合酶链反应(tetraprimer amplif i cation refractory mutation systempolymerase chain reaction,T-ARMS-PCR)[5-6]和温度开关(temperature-switch polymerase chain reaction,TSP)等[7-8]。但目前这些方法的产物往往只能通过溶解曲线和电泳进行检测。这些检测技术不仅需要比较昂贵的仪器,还容易导致扩增产物之间的交叉污染[9-10]。鉴于SNP检测的重要意义,本研究结合聚合酶链反应(polymerase chain reaction,PCR)和位点特异性引物延伸反应(allele-specific extension,ASE)的优点对有关SNP位点的基因型进行检测,探讨PCR-ASE在SNP基因型检测方面的应用可行性。
1 材料和方法
1.1 材料
Taq DNA 聚合酶以及所有的外围扩增引物(PF和PR)、ASE引物和探针均购于生工生物工程(上海)有限公司;引物和探针的有关信息见表1。ASE引物和探针的二级结构以及相互之间的杂交关系可通过Integrated DNA Technologies 公司的引物设计软件(http://eu.idtdna.com/pages/scitools)进行分析,以避免假阳性的出现。在针对不同SNP位点进行检测时,需设计2条PCR外围扩增引物,其退火温度为66~76 °C;相关的ASE引物的5'端需标记异硫氰酸荧光素(fluorescein isothiocyanate,FITC)或者地高辛(digoxin,DIG),其退火温度为45~50 °C。同时为了增加ASE引物的反应特异性,可将SNP位点设计在ASE引物3'端的倒数第2位[11-13]。整个PCR-ASE扩增过程在Gene Touch(PCR)基因扩增仪(杭州博日科技有限公司)上进行。Nanodrop 2000微量分光光度计购自美国Thermo Fisher Scientific有限公司。PCR-ASE产物检测所需的核酸试纸条和全封闭式靶核酸扩增物检测装置均购自杭州优思达生物技术有限公司。
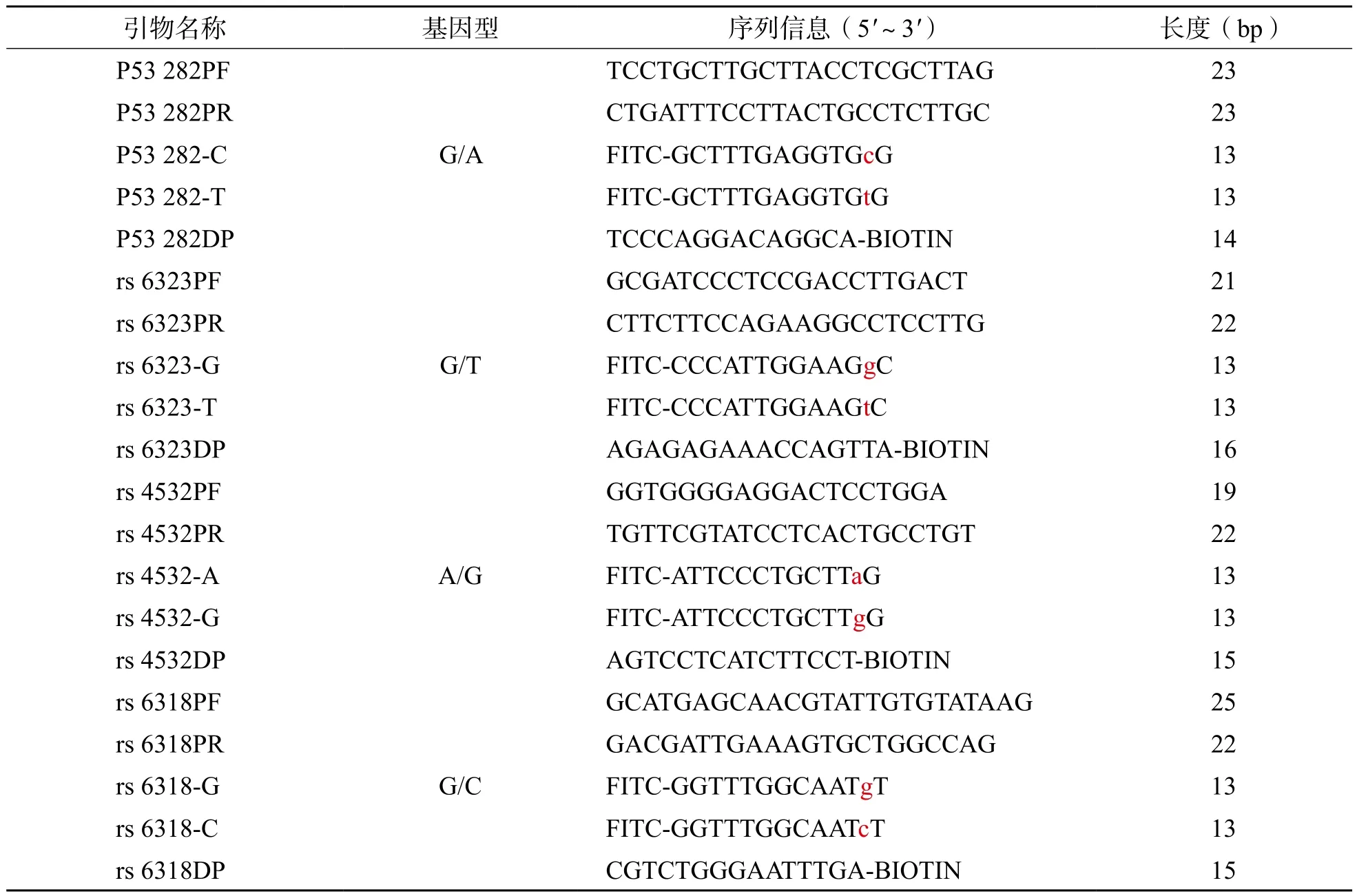
表1 本研究所使用的引物、探针及相关序列
1.2 方法
1.2.1 血液中人基因组DNA的提取 对由杭州艾迪康医学检验中心在2016年9—12月所采集的来自19名不同志愿者的全血样本(1 mL),先采用蛋白酶K在56 ℃进行过夜消化处理;再采用酚氯仿抽提法提取人类基因组DNA。所提取的人类基因组DNA可先通过Nanodrop 2000微量分光光度计对其浓度进行测量,然后放置在-80 ℃进行长期保存。
1.2.2 PCR-ASE和检测 PCR-ASE主要分为4个步骤:PCR扩增、ASE延伸、ASE延伸产物杂交和核酸试纸条检测。(1)通过PCR外围扩增引物对包含SNP位点的靶模板在较高退火温度的条件下进行指数扩增(扩增步骤1),从而达到富集靶模板的目的;(2)在较低退火温度的条件下,只有当ASE引物与SNP位点基因型一致时,才能在DNA聚合酶的作用下进行线性扩增(扩增步骤2),从而达到特异性检测的目的;(3)ASE引物延伸所产生的单链产物可以与体系中的探针进行杂交,进而形成双标记杂交产物;(4)形成的双标记杂交产物可以通过核酸试纸条进行可视化检测。见图1。PCR-ASE的总反应体积为20 μL,成分分别为50 mmol/L KCl、10 mmol/L Tris HCl(pH值7.6)、1% Triton X-100;dNTP(2 mmol/L);Mg2+3 mmol/L;Taq DNA聚合酶 1 U;模板DNA 3 μL;外围扩增引物的浓度为0.2 μmol/L,而ASE引物和探针的浓度均为 0.1 μmol/L。PCR-ASE程序为2个扩增步骤: 95 ℃ 5 min;然后在94 ℃ 30 s;66 ℃30 s的条件下运行40个循环(扩增步骤1);接着在94 ℃ 30 s;50 ℃ 30 s的条件下运行5个循环(扩增步骤2);最后在95 ℃放置5 min。所得的反应产物按照核酸试纸条或全封闭式靶核酸扩增物检测装置所提供的说明书进行检测和判读。如果核酸试纸条的质控线(生物素标记)和检测线(抗FITC或抗DIG抗体)上同时出现红色线条则为阳性;如果只有质控线上出现红色线条则为阴性;如果质控线和检测线上均无条带出现则认为该次检测无效。在PCR-ASE中,5'端FITC或DIG 标记的ASE探针会在DNA聚合酶的作用下进行延伸(扩增步骤2)。而延伸的单链产物会与3'端生物素标记的探针进行结合,形成同时包含生物素和FITC或DIG的双标记产物。在检测过程中,该双标记产物会由于生物素与亲和素之间的作用,先与试纸条加样端上的亲和素标记的红色颗粒结合从而形成红色颗粒-扩增产物-FITC或DIG复合物,并最终会由于FITC和抗FITC抗体或DIG和抗DIG抗体之间的相互作用而被固定在检测线上,并使检测线显示出红色条带。而当无扩增产物形成时,亲和素标记的红色颗粒将移动至质控线,并与质控线上的生物素进行结合。
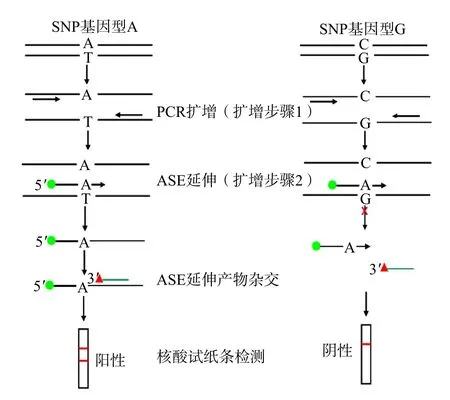
图1 PCR-ASE的原理图
1.2.3 多重PCR-ASE 在PCR-ASE中分别加入2种分别标记FITC和DIG的针对不同SNP基因型的ASE引物,然后按照1.2.2所述的条件进行反应。所得的扩增产物需用2条检测线上分别标记了抗FITC抗体和抗DIG抗体的核酸试纸条进行检测。在进行多重检测时,所采用的核酸试纸条可不包含质控线。SNP基因型的检测结果可根据不同检测线上的抗体类型进行判读。
1.2.4 基因测序和敏感性检测 为了检测PCRASE的反应敏感性,先通过Nanodrop 2000微量分光光度计对人类基因组DNA(纯合子)浓度进行测量,然后对测量后的基因组DNA进行10倍系列的浓度梯度稀释,稀释得到的不同浓度梯度的样本再分别通过含不同ASE引物的PCR-ASE进行检测。同时,本研究基因测序方法被用来验证PCR-ASE检测结果的准确性。
2 结果
2.1 不同扩增步骤循环数对PCR-ASE的影响
由于PCR-ASE中包含2个扩增过程,而2个扩增过程的目的存在很大的区别。因此,为了验证2个扩增步骤中循环数的不同对结果的影响,本研究对2个扩增步骤中的循环数进行了相应的分配,然后用不同ASE引物(A型和G型)对同一个纯合子样本(G型)进行检测,见图2。图2中扩增步骤1的循环数从20逐步增加到45,而步骤2的循环数则从25逐步减少到0,而总的循环数一直保持在45。根据结果显示,总体来说随着扩增步骤2数量的减少,来自于A型ASE引物所导致的非特异性信号逐步减弱并最终消失。当扩增步骤2的循环数>15时,A型ASE引物存在明显的非特异性信号。而当扩增步骤2少于10个循环时,只有与模板基因型一致的G型ASE引物才出现阳性信号。但是当扩增步骤2为0个循环时,2种ASE引物的检测结果均为阴性。
2.2 多重PCR-ASE检测

图2 PCR-ASE中2个不同扩增步骤数量对结果的影响
对多重PCR-ASE所得的扩增产物需用不同检测线上分别标记了抗FITC抗体和抗DIG抗体的核酸试纸条进行检测,检测结果见图3。图3分别显示了p53基因282位点3种不同基因型的检测结果G/G、A/A、G/A。如果只有上面或者下面1条检测线出现红色条带则分别代表该样本为纯合子G和纯合子A,而2条检测线上均出现条带的样本则为杂合子。见图3。
2.3 PCR-ASE敏感性检测和准确性的验证
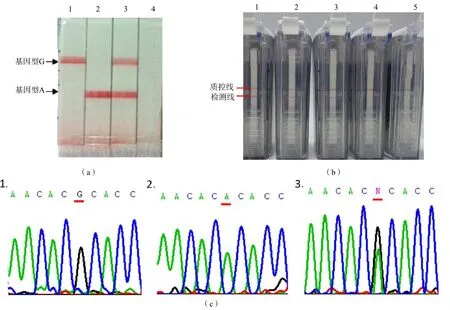
图3 多重PCR-ASE和敏感性检测结果
对已知浓度的纯合子人类基因组DNA进行10倍系列的浓度梯度稀释,然后通过相应的PCR-ASE对所得的样本进行检测,结果见图3。随着样本浓度的降低,检测线上的信号强度逐渐减弱,PCR-ASE的反应敏感性为88 ng/反应。而当ASE引物与模板基因型不完全配对时,该梯度稀释的结果全部为阴性。
为了验证PCR-ASE检测结果的准确性,分别对19份临床样本的5个不同的SNP位点进行检测,并将其检测结果与基因测序法进行比较,符合率为100%。见图3。
3 讨论
SNP是第3代遗传标记和目前生物学研究的热点,随着人类基因组学、药物基因组学以及精准医疗的发展,如何快速地对其进行有效的检测就显得尤为迫切[14]。目前SNP的检测技术主要有基因测序、PCR-限制性片段长度多态性、高效液相、基因芯片等[14],但是这些方法相对来说操作比较复杂,技术要求高,仪器设备昂贵。这些特点在一定程度上限制了SNP检测的应用,特别是在一些发展中国家中的使用。为了解决这一问题,我们开发了一种操作简单和技术要求低的基于PCR-ASE的SNP检测方法,其结果可通过核酸试纸条进行可视化检测。
PCR-ASE原理分别结合了PCR快速有效扩增靶基因和AS-PCR能对SNP不同基因位点进行区分的优点。PCR-ASE先通过PCR外围扩增引物对包含SNP位点的靶模板在较高退火温度的条件下进行指数扩增(扩增步骤1),从而达到富集靶模板的目的。而在此过程中,ASE引物由于其退火温度较低而无法参与到扩增步骤1中。然后在较低退火温度的条件下,在已扩增的靶模板基础上,只有当ASE引物与SNP位点基因型一致时,其才能在DNA聚合酶的作用下进行线性扩增(扩增步骤2),从而达到特异性检测的目的。在扩增步骤2中,由ASE引物延伸所产生的单链产物可以与体系中的探针进行杂交,进而形成双标记杂交产物。而该双标记杂交产物可以通过核酸试纸条进行可视化检测。
为了验证PCR-ASE中的扩增步骤1和扩增步骤2对检测结果的影响,在维持总循环数不变的情况,逐步改变扩增步骤1和扩增步骤2 中的扩增循环数的分布。在扩增步骤1循环数较少的情况下会导致有大量的外围扩增引物残留,而残留的外围扩增引物就会参与到扩增步骤2中。根据以往的文献,由于Taq DNA 聚合酶保真度的问题,在退火温度较低的情况下,AS-PCR扩增过程存在一定的出错概率[13],因此非特异性ASE引物在与模板不完全互补的情况下也可以进行结合并延伸,进而产生非特异性模板。在步骤2循环数较多的情况下,该非特异性模板会在残留的外围扩增引物与非特异性ASE引物的共同作用下进行指数型扩增,最终导致假阳性的出现。但是随着扩增步骤1循环数的增加,外围扩增引物会被大量消耗。因此在进行扩增步骤2时,可有效避免外围扩增引物对扩增步骤2的干扰,ASE引物只能进行单链延伸反应,只起到一个线性扩增的效应,而无法对非特异性信号进行有效的放大。同时随着扩增步骤2循环数的减少,可进一步抑制非特异性信号。但是如果将扩增步骤2循环数降低为零时,由于ASE引物无法参与到扩增步骤1中,就会导致结果的假阴性。
为了降低PCR-ASE操作的复杂性,p53基因282位点3种不同基因型模板被用来对多重PCR-ASE的可行性进行验证。当模板为杂合子时,2条检测线均出现了红色条带;而当模板基因型为纯合子时,只有1条检测线出现信号。通过对已知浓度的人基因组DNA进行10倍系列的浓度梯度稀释,然后利用PCR-ASE进行检测,结果显示PCR-ASE的敏感性为88 ng/反应。同时为了验证PCR-ASE的准确性,分别对19份临床样本的5个不同的SNP位点进行基因检测,其检测结果与PCR-ASE结果完全一致。这些结果均证明了PCR-ASE是一种简单、快速、结果准确可靠、适合于普通实验室开展的SNP基因型检测新方法。
[1] MÉSZÁROS B,ZEKE A,REMÉNYI A,et al.Systematic analysis of somatic mutations driving cancer:uncovering functional protein regions in disease development[J]. Biol Direct,2016,11:23.
[2] ROSES A D. Pharmacogenetics and the practice of medicine [J]. Nature,2000,405(6788):857-865.
[3] BARBANO R,PASCULLI B,COCO M,et al.Competitive allele-specific TaqMan PCR(Cast-PCR) is a sensitive,specific and fast method for BRAF V600 mutation detection in Melanoma patients[J]. Sci Rep,2015,5:18592.
[4] NAFA K,HAMEED M,ARCILA M E. Locked nucleic acid probes(LNA) for enhanced detection of low-level,clinically significant mutations[J].Methods Mol Biol,2016,1392:71-82.
[5] JUNG H,NAM H,SUH J G. Rapid and efficient identification of the mouse leptin receptor mutation(C57BL/KsJ-db/db) by tetra-primer amplification refractory mutation system-polymerase chain reaction(ARMS-PCR) analysis [J]. Lab Anim Res,2016,32(1):70-73.
[6] NEWTON C R,GRAHAM A,HEPTINSTALL L E,et al. Analysis of any point mutation in DNA. The amplification refractory mutation system(ARMS)[J]. Nucleic Acids Res,1989,17(7):2503-2516.
[7] THANH LE P,KHOO K. Temperature switch PCR(TSP):a gel-based molecular marker technique for investigating single nucleotide polymorphisms[J].Methods Mol Biol,2014,1145:37-46.
[8] TABONE T,MATHER D E,HAYDEN M J.Temperature switch PCR(TSP):robust assay design for reliable amplification and genotyping of SNPs[J]. BMC Genomics,2009,10:580.
[9] PAPP A C,PINSONNEAULT J K,COOKE G,et al. Single nucleotide polymorphism genotyping using allele-specific PCR and fluorescence melting curves[J]. Biotechniques,2003,34(5):1068-1072.
[10] MEDINTZ I,WONG W W,BERTI L,et al.High-performance multiplex SNP analysis of three hemochromatosis-related mutations with capillary array electrophoresis microplates[J]. Genome Res,2001,11(3):413-421.
[11] IMYANITOV E N,BUSLOV K G,SUSPITSIN E N,et al. Improved reliability of allele-specific PCR[J]. Biotechniques,2002,33(3):484.
[12] LATORRA D,CAMPBELL K,WOLTER A,et al.Enhanced allele-specific PCR discrimination in SNP genotyping using 3'locked nucleic acid(LNA)primers[J]. Hum Mutat,2003,22(1):79-85.
[13] NASIS O,THOMPSON S,HONG T,et al.Improvement in sensitivity of allele-specific PCR facilitates reliable noninvasive prenatal detection of cystic fi brosis[J]. Clin Chem,2004,50(4):694-701.
[14] DEND Q,RAMSKÖLD D,REINIUS B,et al.Single-cell RNA-seq reveals dynamic,random monoallelic gene expression in mammalian cells[J].Science,2014,343(6167):193-196.
猜你喜欢
时代汽车(2021年18期)2021-09-17 11:51:59
电动工具(2020年6期)2020-12-29 05:53:36
设备管理与维修(2020年2期)2020-03-24 13:12:06
中国医疗保险(2017年5期)2017-05-17 08:26:39
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
中国康复理论与实践(2015年10期)2015-12-24 05:42:46
现代电生理学杂志(2015年1期)2015-07-18 11:02:16
现代检验医学杂志(2015年6期)2015-02-06 01:44:02
实验动物与比较医学(2014年5期)2014-02-28 14:53:10
中国糖料(2013年1期)2013-01-22 12:28:23
