
重氮化合物与富电子烯烃的环加成反应研究进展
2018-07-05 10:35何龙,胡方,曹菲
西华师范大学学报(自然科学版) 2018年2期
何 龙,胡 方,曹 菲
(西华师范大学 化学化工学院,四川 南充 637009)
自19世纪Curtiu首次报道α-重氮化合物(重氮乙酸乙酯)以来[1-2],α-重氮化合物已经成为一类用途广泛的有机合成中间体,被应用于各种有机合成反应中[3-4]。重氮在光[5]、热、金属以及手性金属配合物的作用下能够分解形成高度活泼的自由基卡宾或金属卡宾中间体。而金属卡宾由于金属的配位使得二价碳中间体的活性降低,因此金属卡宾相对于传统的自由基卡宾而言更加稳定,具有良好的可控性等优点;金属卡宾的这些优势使得其在有机化学合成中越来越得到重视[6]。
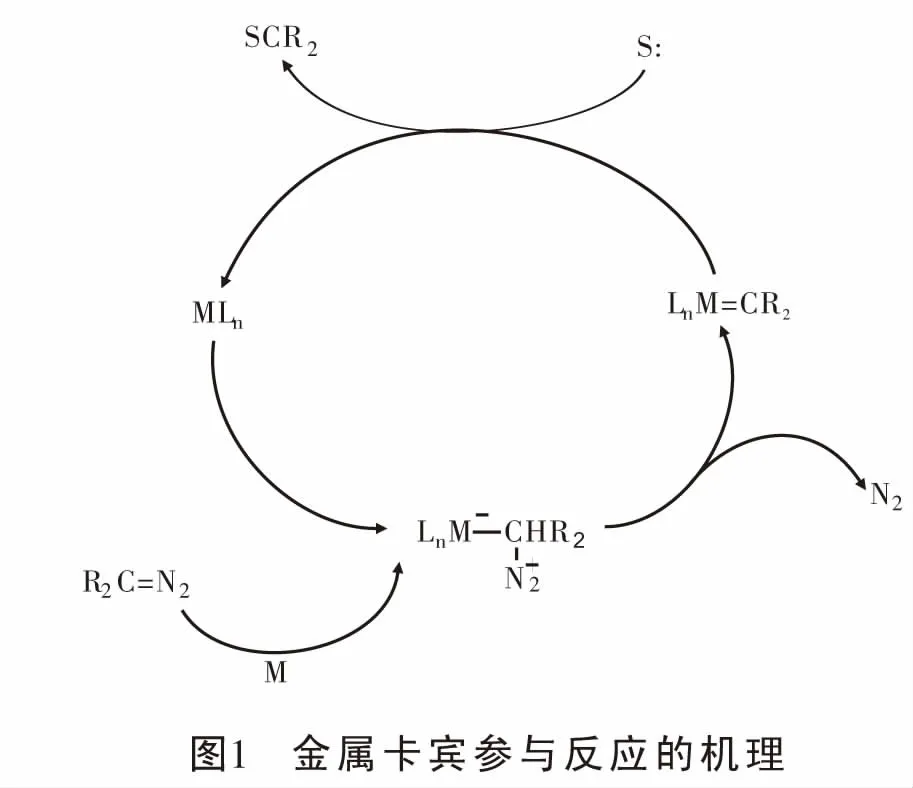
重氮化合物形成的金属卡宾参与的反应中,由于其自身的特点,它既可以作为亲核试剂[7-10]、也可以作为亲电试剂。金属催化重氮分解形成的金属卡宾作为亲电试剂参与反应的机理为:首先金属对重氮化合物亲电加成形成相对稳定的金属卡宾,然后产生的卡宾迁移到富电子底物上并重新生成具有催化活性的金属催化剂MLn完成循环[11](图1)。近年来,关于重氮形成的金属卡宾作为亲电试剂参与的反应,主要报道了它与含杂原子(氧、硫、氮等)的富电子体系[12-18]、富电子芳烃[19-20]以及富电子共轭二烯等的反应。因富电子共轭二烯与重氮形成的金属卡宾参与的反应展示了自由基化学新的可能性,本文主要介绍近年来过渡金属催化重氮化合物与富电子共轭二烯以及烯烃反应的研究进展, 并对反应机理进行探讨。
1 重氮化合物与富电子共轭烯烃的[2+1]环加成反应
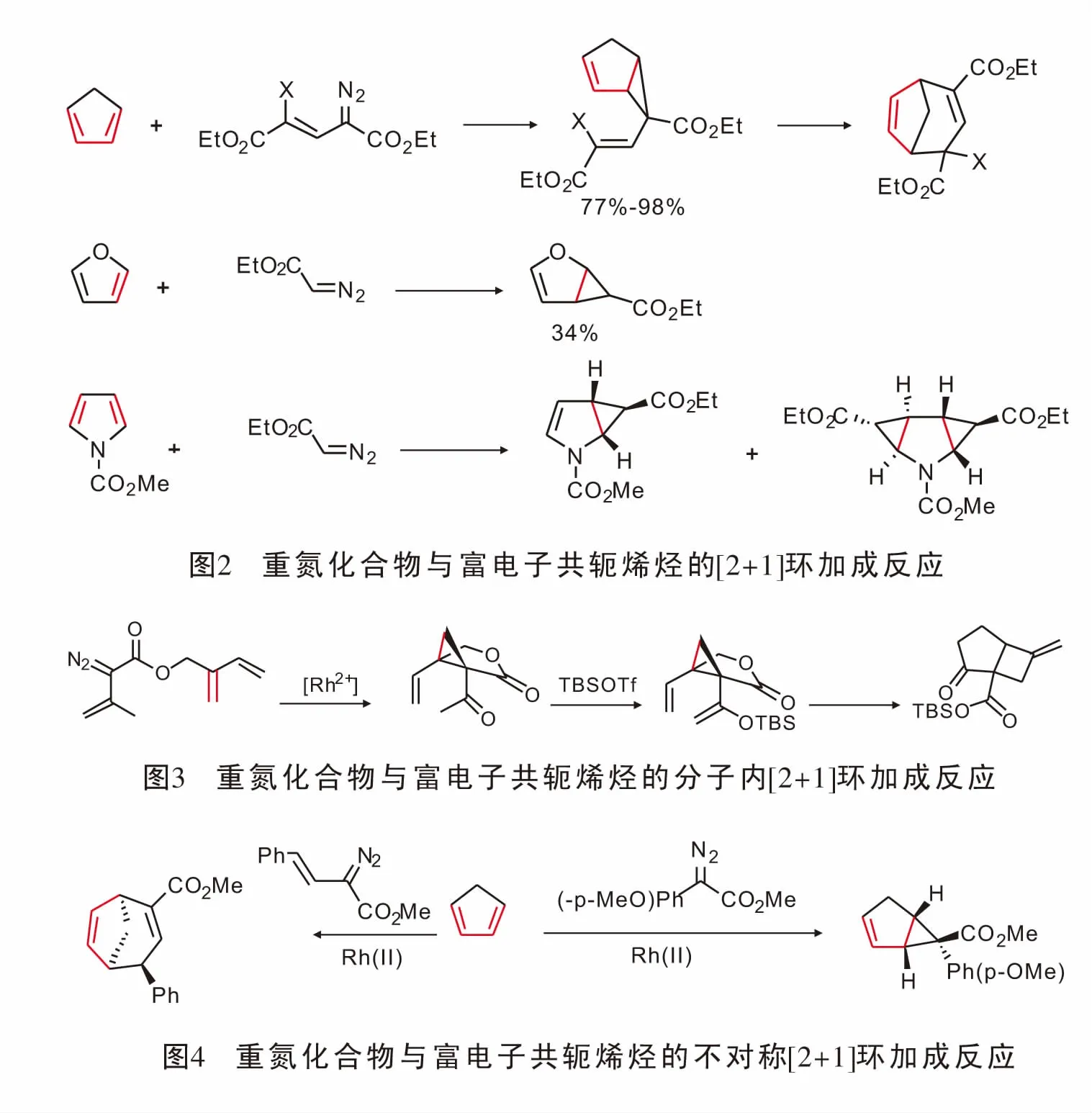
由于富电子二烯含有两个可供金属卡宾反应的双键,因而双键上的电子效应以及空间效应在反应选择性方面起着至关重要的作用;同时由于[2+1]环加成形成三元环产物,环张力大,容易引发一系列的重排,导致其在合成领域备受关注[21]。1988年,Wulff首次报道了重氮化合物与金属铬形成的金属卡宾与环戊二烯的[2+1]环加成反应,以及由三元环在高温下发生扩环的[4+3]产物[22],该反应化学收率可以达到98%,由此化学家们对重氮化合物与环戊二烯及呋喃、吡咯的反应展开了相关研究[23](图2)。
1996年,Davies[24]实现了铑催化重氮化合物与分子内的共轭二烯发生[2+1]环加成反应并在形成[4.3.1]二环化合物后实现了分子内重排;然而该小组并未对反应的不对称催化进行研究(图3)。
2010年,Davies[25]实现了手性羧酸铑催化重氮与环戊二烯的[2+1]环加成反应,反应得到稳定的具有光学活性的二环化合物。在使用α-不饱和重氮得到三元环化合物后,其迅速发生重排生成具有光学活性的桥环化合物(图4)。
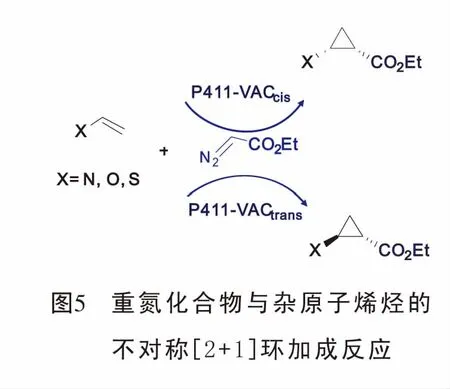
2018年,Arnold[26]教授报道了具有立体选择性的血红素蛋白酶催化杂原子烯烃与重氮化合物的[2+1]环加成反应,合成了杂原子取代苯胺环丙烷结构。该反应的优点在于可以便利的通过定向调节血红蛋白酶,有效的促进新的底物与重氮的环丙烷化且扩大了铁血红素蛋白的催化功能(图5)。
2 不饱和重氮化合物与富电子烯烃的[3+2] 环加成反应
1998年,Davies[27]小组报道了不饱和重氮化合物在金属Rh催化作用下形成金属卡宾,与乙烯基醚发生[2+1]环加成反应得到高选择性的乙烯基环丙烷产物,再在二乙基氯化铝路易斯酸的作用下活化双键发生重排形成环戊烯结构衍生物。2001年,Davies[28]实现了脯氨酸衍生的手性羧酸铑直接催化不饱和烯丙基重氮化合物与双键生成具有三个手性中心的不饱和五元环结构,并证明了烯丙基对重氮形成的金属卡宾具有稳定的作用。其后不饱和重氮化合物与富电子双键的反应一直备受关注。2010年,Park[29]通过铜催化实现重氮化合物与双键反应高产率的构建吡咯环结构。2016年,Doyle[30]课题组报道了烯氨基甲酸酯与亲电金属烯醇类卡宾的[3+2]环加成反应,以高化学收率、高达98% ee的对映选择性和高非对映选择性得到手性不饱和五元环结构化合物(图6)。
吲哚作为重要的有机合成子,在重氮参与的[3+2]环加成领域同样受到高度的重视。Davies[31]发表了α-β不饱和乙烯基重氮化合物与吲哚的不对称[3+2]环加成反应。2016年,Doyle[32]小组报道了Rh催化亲电子烯醇重氮化合物分解形成亲电子烯醇卡宾与吲哚类化合物发生[3+2]环加成反应,以高区域选择性和对映选择性合成了具有手性的环戊烷稠合的吲哚类化合物(图7)。

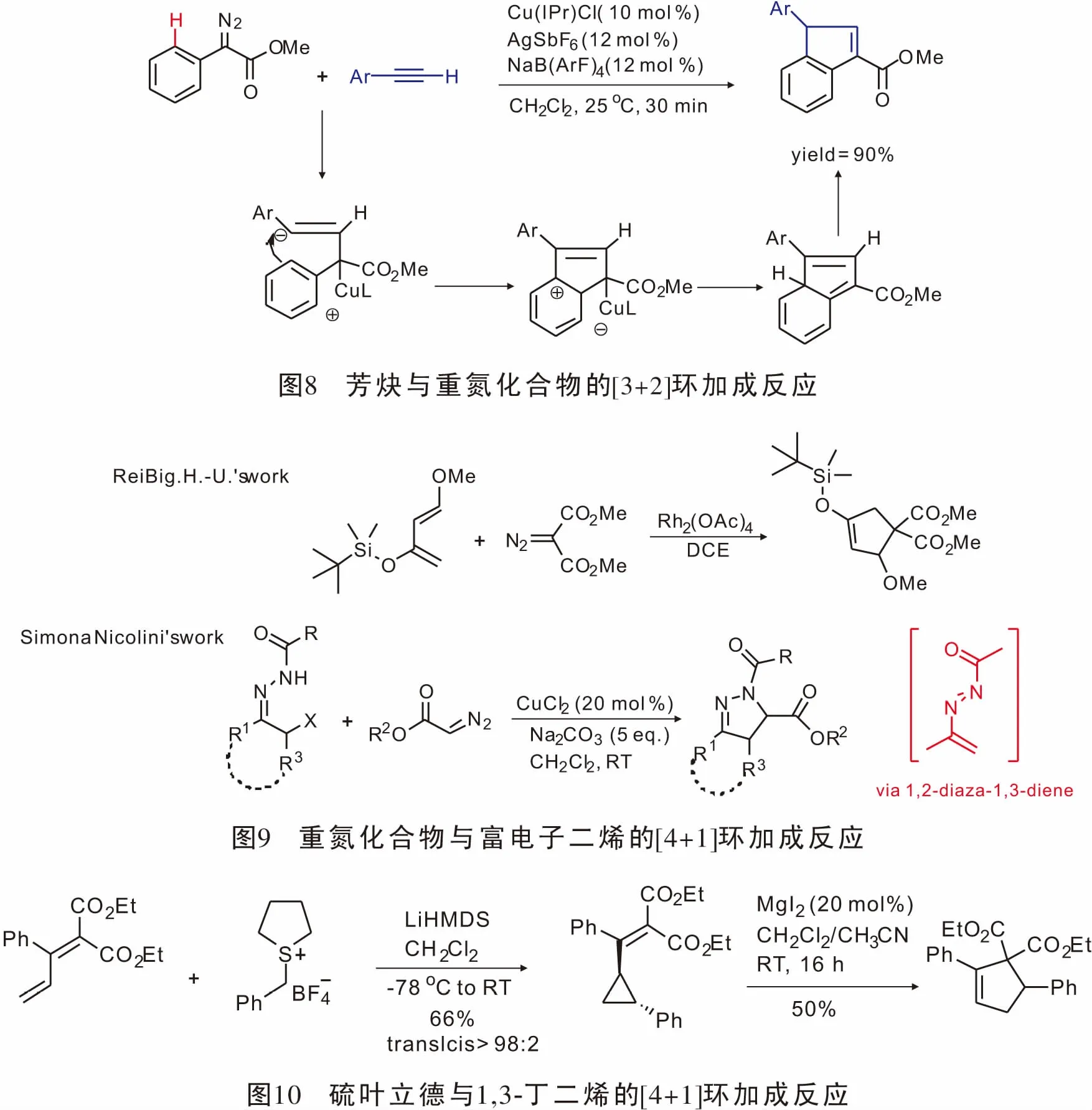
近年来C-H键活化参与的反应得到了大量的发展,在此基础上,2008年,Chang[33]通过sp2C—H键活化以高产率实现了第一例末端炔烃与α-重氮苯乙酸酯参与的[3+2]环加成反应(图8)。
3 重氮化合物与富电子共轭烯烃的[4+1]环加成反应
重氮化合物与过渡金属形成的金属卡宾能够与乙烯基类化合物发生[2+1]环加成反应,且其发展相对成熟;基于这一性质,人们设想重氮化合物形成的金属卡宾能否与富电子共轭二烯以及原位生成类共轭二烯发生[4+1]环加成反应。1996年,ReiBig.H.U.首次实现了重氮化合物与富电子二烯的[4+1]环加成反应[34],最高获得43%的产率,首次实现了重氮形成的金属卡宾与碳原子亲核试剂的[4+1]环加成反应。2014年,Nicolini小组[35]报道了原位生成的1,2-二氮杂-1,3-二烯与重氮酯反应可直接得到单、双、三取代的4,5-二氢吡唑结构(图9)。
2015年,Robiette课题组[36]报道了硫叶立德与1,3-丁二烯发生不对称[4+1]环加成反应。该方法先是具有手性的硫叶立德与1,3-丁二烯反应生成光学活性的乙烯基环丙烷,再在MgI2的作用下扩环得到[4+1]产物。该方法具有良好的官能团兼容性与立体选择性(图10)。
靛红衍生的重氮化合物参与的反应是构建具有季碳中心螺环化合物的有效方法之一,在近几年得到了大多数课题组的青睐。2017年,由Noni教授[37]报道的Rh催化重氮氧化吲哚作为C1合成子与环丁烯酮类化合物反应生成中间体环丙烷乙烯酮结构后;在相对温和的条件下,以较好的收率发生1,3-迁移生成螺环氧化吲哚(图11)。
2017年,Ashfeld教授[38]报道了Rh(II)催化重氮氧化吲哚与类似1,3-二烯结构的化合物2a反应,从而构建含有吡咯酮的螺环氧化吲哚类化合物。文中使用乙烯基异氰酸酯作为1,4-偶极子与金属卡宾形成三元环结构后,在碱的作用下开环形成螺环结构4a (图12)。
2018年,Liu课题组[39]报道了富电子Danishefsky二烯与靛红衍生的重氮在金属铑催化下发生不对称[4+1]环加成反应。此反应能够通过乙烯基环丙烷结构中间体开环,在三氟乙酸的作用下一锅法以较高的产率及良好的对映选择性得到具有一个季碳中心的螺环环戊烯酮类结构,反应对底物具有广普性,同时该反应也为手性金属铑催化的不对称[4+1]环加成反应提供了新的研究思路(图13)。
4 重氮化合物与富电子共轭烯烃的[4+2]环加成反应
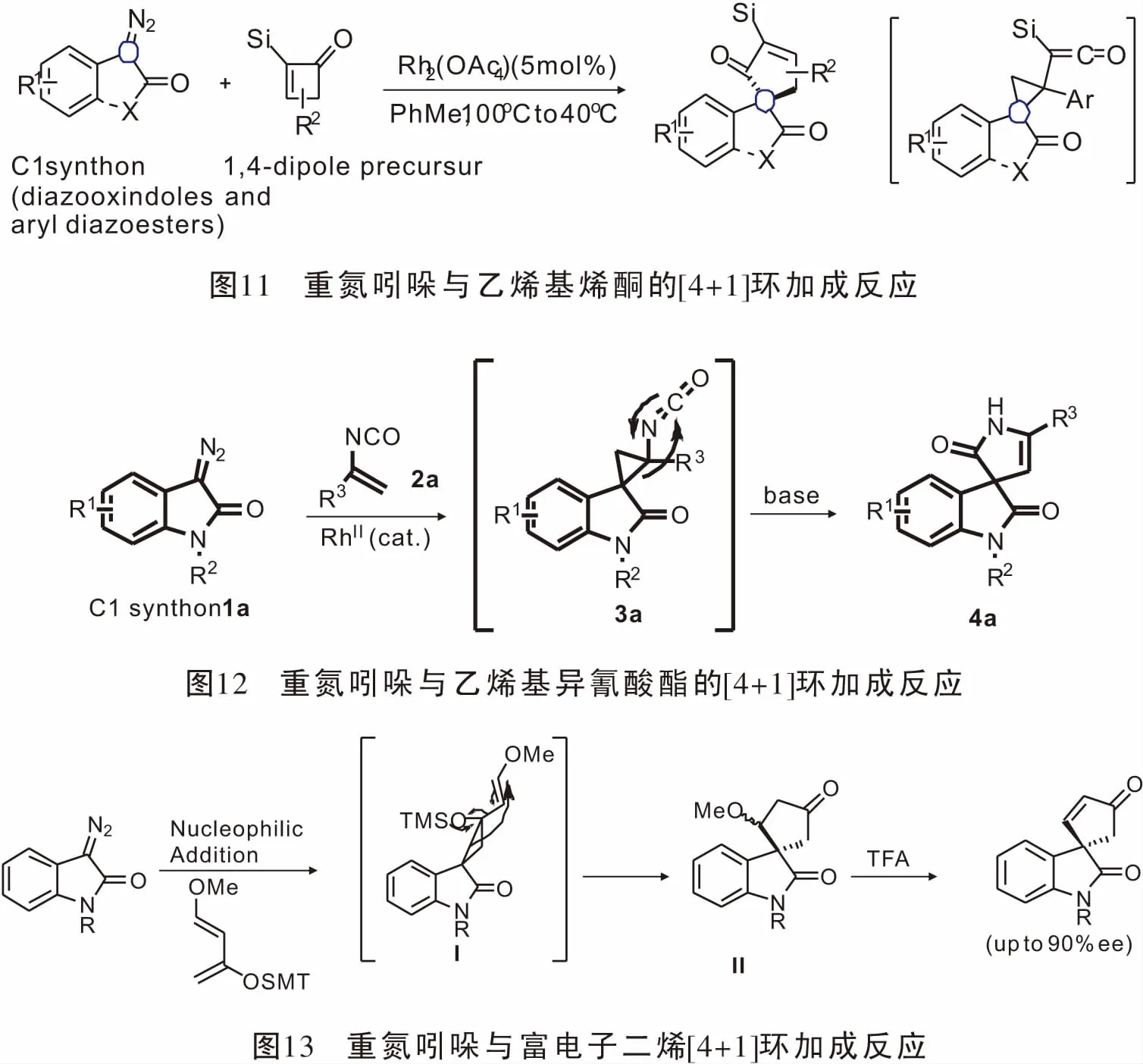
2015年,Zeng小组[40]报道了Rh(III)催化氮酰胺吲哚与α-羰基重氮化合物经sp2C-H活化,羰基直接参与的[4+2]环加成反应(图14),简单高效的合成了2H-嘧啶并[1,6-a]吲哚-1-酮,且具有良好的底物普适性。
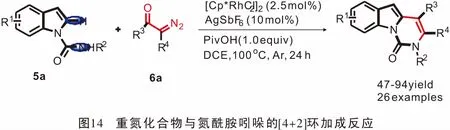
2016年,Doyle团队[41]首次发现了1,3-二烯与重氮酯之间的分子内[4+2]环加成反应(图15)。反应经历Au(I)催化炔丙基重氮重排得到中间体1,3-二烯芳基重氮乙酸酯(Z)-7a;其中间体经过4天的反应后,可以以74%的产率得到[4+2]环加成产物。

5 重氮化合物与富电子共轭烯烃的[4+3]环加成反应
自Davies[42]1985年实现烯丙基重氮形成金属卡宾与共轭二烯构建七元环化合物后,其方法迅速发展。先后实现了烯丙基卡宾与呋喃[43]、环戊二烯[44]、1,3-丁二烯[45]、吡咯[46-47]、苯环[48]以及其分子内的反应[49]构建七元环。2008年Davies[50]课题组实现了苯并呋喃衍生的重氮与富电子共轭二烯的不对称[4+3]环加成反应,并达到了83%的产率,98% ee的对映选择性。同年Davies[51]通过烯丙基卡宾与共轭二烯实现不对称[4+3]环加成反应,同时应用该方法成功的完成(+)-Barekoxide、(-)-Barekol的全合成,使此方法在合成领域大放异彩。2014年,Davies[52]课题组实现了烯醇金属卡宾与富电子二烯的不对称[4+3]环加成反应。反应经历了三元环中间过渡态,然后开环实现不对称[4+3]环加成反应。Davies小组使用实验室发展的手性羧酸铑催化剂实现了99% ee的对映选择性,大于30:1的非对映选择性(图16)。
2015年,Wang课题组[53]报道了Rh催化简单、易得的重氮亚胺与呋喃的反应,合成了吡啶并吲哚以及四氢呋喃并吡咯吲哚类结构。此反应经历了α-亚胺基铑卡宾中间体的反应机理,然后发生消除、构建6富电子环化结构(图17)。
2017年,Wang小组[54]报道了Rh催化重氮亚胺与1,3-二烯的[4+3]环加成反应。以良好的收率实现了铑催化3-重氮二氢吲哚-2-亚胺与1,3-二烯的[4+3]环加成反应,且具有良好的底物适用范围。当使用2-[(三甲基甲硅烷基)氧基]-1,3-丁二烯作为二烯组分,可以构建氮杂并[2,3-b]吲哚-4(H)-酮;而使用1,3-环己二烯或1,3-环戊二烯,则得到相应的[3+2]环加成产物(图18)。
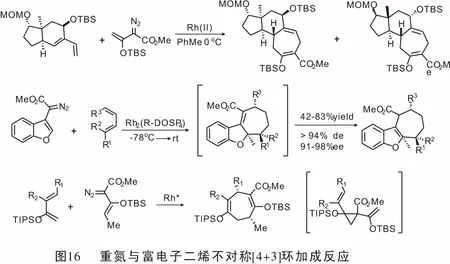

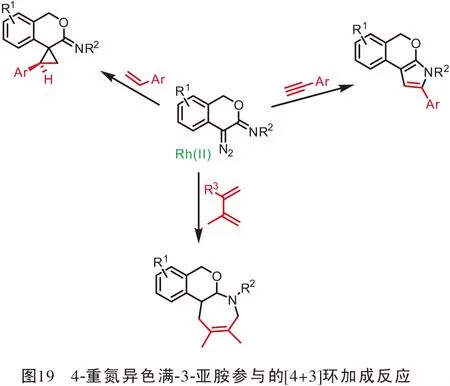
同年,Wang教授[55]研究了4-重氮异色满-3-亚胺作为新型金属卡宾前体的合成应用。在Rh(II)配合物的催化作用下,通过与α-烯烃发生[2+1](即环丙烷化)或与共轭二烯发生[4+3]环加成反应可以得到螺[环丙烷-1,4′-异色满]-3′-亚胺和四氢异色烯并[3,4-b]吖庚因。 当使用Rh(II)/ AgOTf作为辅助催化剂时,发生4-重氮异色满-3-亚胺与末端炔的[3+2]环加成反应,得到2-芳基-3,5-二氢异色烯[3,4-b]吡咯(图19)。
6 结论与展望
综上所述,重氮化合物与金属形成的金属卡宾作为亲电试剂参与的反应以其独特的优势在近几年来越来越受到关注。而金属卡宾作为亲电试剂直接与碳原子亲核试剂参与的反应主要集中在[2+1]、[3+2] 、[4+1]、[4+2]、[4+3]环加成反应上。这些反应为有机合成提供了新的方法学,同时也为铑在不对称催化方法学上的应用展示了新的可能性。然而不饱和重氮经由烯醇金属卡宾与富电子共轭二烯的不对称环加成反应的研究相对较少,因此发展普通的廉价金属与新型手性配体络合催化重氮金属卡宾的不对称反应具有重要的意义。
参考文献:
[1] CURTIUS T.Ueber diazoessigsäure und ihre derivate[J].Adv.Synth.Catal.,1888,38:396-440.
[2] CURTIUS T.Ueber die einwirkung von salpetriger säure auf salzsauren glycocolläther[J].Eur.J.Inorg.Chem.,2010,16(2):2230-2231.
[3] CHE J,XING D,HU W.Metal-Catalyzed cross-coupling of terminal alkynes with different carbene precursors[J].Curr.Org.Chem.,2016,20(1):41-60.
[4] XU X,DOYNE M P.The [3+3]-cycloaddition alternatives for hete-rocycle syntheses:Catalytically generated metalloenolcarbenes as dipolar adducts[J].Acc.Chem.Res.,2014,45(25):1396-1399.
[5] MURATA S,KOBAYASHI J,KONGON C,et al.Photochemistry of 2,4-Bis-(diazo)-1,2,3,4-tetrahydro-naphthalene-1,3-dio-ne:selective photodecomposition of one of the two inequivalent diazo groups[J].J.Am.Chem.Soc.,1998,120(35): 9088-9089.
[6] VIJAY NAIR C,RAJESH A U,VIOND S,et al.Strategies for heterocyclic construction via novel multicomponent reactions based on isocyanides and nucleophilic carbenes[J].Acc.Chem.Res.,2003,36(12):899-907.
[7] HASHIMOTO T,MIYAMOTO H,NAGANAWA Y.Stereoselective synthesis of a-alkyl-b-keto imides via asymmetric redox C-C bond formation between α-alkyl-α-diazocarbonyl compounds and aldehydes[J].J.Am.Chem.Soc.,2010,41(2):2422-2425.
[8] GAO L,KANG B C,RYU D H.Catalytic asymmetric insertion of diazoesters into aryl-CHO bonds:highly enantioselective construction of chiral all-carbon quaternary centers[J].J.Am.Chem.Soc.,2013,135(39):14556-14559.
[9] CHEN X,HU X,BAI S.Rh(III)-catalyzed [4+2] annulation of indoles with diazo compounds:Access to pyrimido [1,6-a] indole-1-(2H)-ones[J].Org.lett.,2015,18(2):192-195.
[10] QIU H,SRINIVAS H D,ZAVALIJ P Y,et al.Unprecedented intramolecular [4+2]-cycloaddition between a 1,3-diene and a diazo ester[J].J.Am.Chem.Soc.,2016,47(31):1808-1811.
[11] DAVIES H M L,BECKWITH R E J.Catalytic enantioselective C-H activation by means of metal-carbenoid-induced C-H insertion [J].Chem.Rev.,2003,103:2861-2904.
[12] BULUGAHAPITIYA P,LANDAIS Y,PARRA-PAPADO L.A stereospecific access to allylic systems using rhodium (II) -vinyl carbenoid insertion into Si-H,O-H,and N-H bonds[J].J.Org.Chem.,1997,62(6):1630-1641.
[13] MAIER T C,FU G C.Catalytic enantioselective O-H insertion reactions[J].J.Am.Chem.Soc.,2006,128(14):4594-4595.
[14] ZHU S F,CHEN C,CAI Y.Catalytic asymmetric reaction with water: enantioselective synthesis of α-hydroxyesters by a copper-carbenoid O-H insertion reaction[J].Angew.Chem.Int.Ed.,2008,47(5):932-934.
[15] HUANG H,HU W.Rhodium-catalyzed reaction of diazoacetates,thiols and azodicarboxylates: an unusual 1,2-aza shift from a sulfonium ylide[J].Synlett.,2007,38(38):1314-1316.
[16] XU B,ZHU S F,ZHANG Z C.Highly enantioselective S-H bond insertion cooperatively catalyzed by dirhodium complexes and chiral spiro phosphoric acids[J].Chem.Sci.,2014,45(32):1442-1448.
[17] YADAGIRI D,ANBARASAN P.Tandem 1,2-sulfur migration and (aza)-diels-alder reaction of β-thio-α-diazoimines: Rhodium catalyzed synthesis of (fused)-polyhydropyridines,and cyclohexenes[J].Chem.Sci.,2015,6(10):5847-5852.
[18] XU X,ZAVALIJ P Y,HU W.Vinylogous reactivity of enoldiazoacetates with donor-acceptor substituted hydrazones:synthesis of substituted pyrazole derivatives[J].J.Org.Chem.,2013,78(4):1583-1588.
[19] GULEVICH A V,GEVORGYAN V.Versatile reactivity of rhodium-iminocarbenes derived from N-sulfonyl triazoles[J].Angew.Chem.Int.Ed.,2013,52(5):1371-1373.
[20] XING D,JING C,LI X.Highly efficient synthesis of mixed 3,3′-Bisindoles via Rh(II)-catalyzed three-component reaction of 3-diazooxindoles with indoles and ethyl glyoxylate[J].Org.Lett.,2013,15(14):3578-3581.
[21] DAVIESAVIES H M L.Tandem cyclopropanation/cope rearrangement:a general method for the construction of seven-membered rings [J].Tetrahedron.,1993,24(43):5203-5223.
[22] WULFF W D,YANG D C,MURRAY C K.Cyclopropanations and cycloadditions of transition metal carbene complexes[J].Cheminform.,1988,60(1):137-144.
[23] DAVIES H M L,ANTOULINAKIS E G.Intermolecular metal-catalyzed carbenoid cyclopropanations[J].Organic Reactions.,2004,34(34):55-57.
[24] DAVIES H M L,CALVO R,AHMED G.Type II intramolecular annulations between vinylcarbenoids and furans[J].Tetrahedron.Lett.,1997,38:1737-1740.
[25] PELPHREY P,HANSEN J,DAVIES H M L.Solvent-free catalytic enantioselective C-C bond forming reactions with very high catalyst turnover numbers [J].Chem.Sci.,2010,41(49):254-257.
[26] BRANDENBERG O F,PRIER C K,CHEN K,et al.Stereoselective enzymatic synthesis of heteroatom substituted Cyclopropanes[J].Acs Catalysis.,2018,8:2629-2634.
[27] DAVIES H M L,KONG N,CHURCHILL M R.Asymmetric synthesis of cyclopentenes by [3+2] annulation between vinylcarbenoids and vinyl Ethers [J].J.Org.Chem.,1998,63:6586-6589.
[28] DAVIES H M L,XIANG B,KONG N,et al.Catalytic asymmetric synthesis of highly functionalized cyclopentenes by a [3+2] cycloaddition[J].J.Am.Chem.Soc.,2001,123(30):7461-7462.
[29] LOURDUSAMY E,YAO L,PARK C M.Stereoselective synthesis of α-diazo oxime ethers and their application in the synthesis of highly substituted pyrroles through a [3+2] Cycloaddition [J].Angew.Chem.Int.Ed.,2010,49(43):7963-7967.
[30] DENG Y,YGLESIAS M V,ARMAN H,et al.Catalytic asymmetric synthesis of cyclopentyl β-amino esters by [3+2] cycloaddition of enecarbamates with electrophilic metalloenolcarbene intermediates[J].Angew.Chem.Int.Ed.,2016,55(34):10108-10112.
[31] LIAN Y,DAVIES H M L.Rhodium-catalyzed [3+2] annulation of indoles [J].J.Am.Chem.Soc.,2009,132(2):440-441.
[32] JING C,CHENG Q Q,DENG Y,et al.Highly regio-and enantioselective formal [3+2]-annulation of indoles with electrophilic enol carbene intermediates [J].Org.Lett.,2016,18(18):4550-4553.
[33] PARK E J,KIM S H,CHANG S.Copper-catalyzed reaction of α-aryldiazoesters with terminal alkynes: a formal [3+ 2] cycloaddition route leading to indene derivatives [J].J.Am.Chem.Soc.,2008,130(51):17268-17269.
[34] SCHNAUBELTRLL,ELKE MARKS,HANS-ULRICH REIBIG,et al.[4+1] Cycloadditions of the rhodium di(methoxycarbonyl) carbenoid to 2-siloxy-1,3-dienes [J].Chem Ber.,1996,129(1):73-75.
[35] ATTANASI O A,CRESCENTINI L D,FAVI G,et al.Interceptive [4+1] annulation of in situ generated 1,2-diaza-1,3-dienes with diazo esters:direct access to substituted mono-,bi-,and tricyclic 4,5-dihydropyrazoles[J].J.Org.Chem.,2014,79(17):8331-8338.
[36] ROUSSEAU O,DELAUNAY T,DEQUIREZ G,et al.Formal asymmetric [4+1] annulation reaction between sulfur ylides and 1,3-Dienes [J].Chem.Eur.J.,2015,21(37):12899-12902.
[37] RODRIGUEZ K X,KALTWASSER N,TONI T A,et al.Rearrangement of an intermediate cyclopropyl ketene in a RhII-catalyzed formal [4+1]-cycloaddition employing vinyl ketenes as 1,4-dipoles and donor-acceptor metallocarbenes[J].Org.Lett.,2017,19(10):2482-2485.
[38] MELOCHE J L,ASHFELD B L.A rhodium(II)-catalyzed formal [4+1]-cycloaddition toward spirooxindole pyrrolone construction employing vinyl isocyanates as 1,4-dipoles [J].Angew.Chem.Int.Ed.,2017,56(24):6604-6608.
[39] CAO F,HU F,LUO G Y,et al.Rhodium catalyzed enantioselective formal [4 +1] cycloaddition of diazooxindoles and the danishefsky diene [J].Asian J.Org.Chem.,2018,7:363-366.
[40] CHEN X,HU X W,SIYI B,et al.Rh(III)-catalyzed [4 + 2] annulation of indoles with diazo compounds:access to pyrimido[1,6 a]indole-1(2H) ones [J].Org.Lett.,2016,18(2):192-195.
[41] ABING DUAN,YU P Y,MICHAEL P,et al.Diazo esters as dienophiles in intramolecular [4+2] cycloadditions:computational explorations of mechanism [J].J.Am.Chem.Soc.,2017,139:2766-2770.
[42] DAVIES H M L,CLARK D M,SMITH T K.[3+ 4] cycloaddition reactions of vinyl carbenoids with furans [J].Tetrahedron.Lett.,1986,17(8):5659-5662.
[43] DAVIES H M L,SMITH H D,KORKOR O.Tandem cyclopropanation cope rearrangement sequence:Stereospecific [3+4] cycloaddition reaction of vinylcarbenoids with cyclopentadiene [J].Tetrahedron.Lett.,1987,18(44):1853-1856.
[44] DAVIES H M L,STAFFORD D G,DOAN B D,et al.Tandem asymmetric cyclopropanation/cope rearrangement.a highly diastereoselective and enantioselective method for the construction of 1,4-cycloheptadienes[J].J.Am.Chem.Soc.,1998,29(31):3326-3331.
[45] DAVIES H M L,CALVO R L,TOWNSEND R J,et al.An exploratory study of type [3+ 4] cycloadditions between vinylcarbenoids and dienes[J].J.Org.Chem.,2000,65(14):4261-4268.
[46] DAVIES H M L,MATASI J J,THORNLEY C.Control of carbenoid reactivity through neighboring group participation [J].Tetrahedron.Lett.,1995,36:7205-7208.
[47] DAVIES H M L,MATASIJ J,HODGES L M,et al.Enantioselective synthesis of functionalized tropanes by rhodium (II) carboxylate-catalyzed decomposition of vinyldiazomethanes in the presence of pyrroles[J].J.Org.Chem.,1997,28 (26):1095-1105.
[48] DAVIES H M L,SMITH H D,HU B,et al.Divergent reaction pathways between rhodium(II)-stabilized vinylcarbenoids and benzenes [J].J.Org.Chem.,1992,57(25):6900-6903.
[49] DAVIES H M L,DOAN B D.Asymmetric synthesis of the tremulane skeleton by a tandem cyclopropanation cope rearrangement [J].Tetrahedron.Lett.,1996,27(39):3967-3970.
[50] OLSON J P,DAVIES H M L.Asymmetric [4+3] cycloadditions between benzofuranyldiazoacetates and dienes: formal synthesis of (+)-frondosin B [J].Org.Lett.,2008,10(4):573-576.
[51] LIAN Y,MILLER L C,BORN S,et al.Catalyst-controlled formal [4+ 3] cycloaddition applied to the total synthesis of (+)-barekoxide and (-)-barekol[J].J.Am.Chem.Soc.,2010,132(35):12422-12425.
[52] GUZMAN P E,LIAN Y,DAVIES H M L.Reversal of the regiochemistry in the rhodium-catalyzed [4+3] cycloaddition between vinyldiazoacetates and dienes [J].Angew.Chem.Int.Ed.,2014,53(48):13083-13087.
[53] WANG C,ZHANG H,LANG B,et al.Rh-catalyzed reactions of 3-diazoindolin-2-imines:synthesis of pyridoindoles and tetrahydrofuropyrroloindoles [J].Org.Lett.,2015,17(18):4412-4415.
[54] LANG B,ZHU H,WANG C,et al.Rhodium-catalyzed cycloadditions between 3-diazoindolin-2-imines and 1,3-dienes [J].Org.Lett.,2017,19(7):1630-1633.
[55] REN A,LANG B,LIN J,et al.4-diazoisochroman-3-imines:a Class of metal carbene precursors for the synthesis of isochromene derivatives[J].J.Org.Chem.,2017,82:10953-10959.
猜你喜欢
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年6期)2021-12-21
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
昆明医科大学学报(2020年12期)2021-01-26
河北理科教学研究(2020年1期)2020-07-24
应用数学(2020年2期)2020-06-24
第一财经(2019年8期)2019-08-26
山东化工(2019年11期)2019-06-26
中南民族大学学报(自然科学版)(2019年1期)2019-04-04
安徽医科大学学报(2015年9期)2015-12-16
