
抗SRP抗体阳性和抗PM/Scl抗体阳性多发性肌炎1例并文献复习
2018-07-02 03:52:54曾艳平柳胤解燕春董红娟初红卢祖能
神经损伤与功能重建 2018年6期
曾艳平,柳胤,解燕春,董红娟,初红,卢祖能
多发性肌炎(polymyositis,PM)是以四肢近端肌肉受累为主要临床表现的获得性肌肉疾病,与皮肌炎、散发性包涵体肌炎及免疫介导坏死性肌病合称为特发性炎性肌病[1,2]。大多数特发性炎性肌病患者血清中可存在肌炎抗体,包括抗信号识别颗粒抗体(抗SRP抗体),有学者提出抗SRP抗体为特异性免疫介导坏死性肌病的标志性抗体[3-5]。本研究报道的1例多发性肌炎患者也存在抗SRP抗体阳性,曾被误诊为免疫介导的坏死性肌病,此病例特殊且少见。
1 临床资料
患者男,65岁,因“渐起双下肢无力2年余,加重半月”于2017年4月22日入院。患者2014年底无明显诱因出现双下肢疼痛无力,外院肌酸激酶8193 U/L,考虑炎性肌病,给予甲强龙冲击治疗,出院后口服强的松维持。期间病情加重2次,均行甲强龙冲击治疗,具体不详。近1个月患者自感症状加重,主要表现为活动后心慌、喘气,上楼梯困难,蹲起费力,门诊肌酸激酶5131 U/L,肌红蛋白>1200 ng/mL,肌肉MRI提示双侧大腿各组肌群肿胀,异常信号。既往有高血压病史10余年,口服硝苯地平缓释片和倍他乐克片,自起病来精神稍差,饮食睡眠可,二便正常,体重下降约10 kg。
入院查体:体温37℃,血压140/91 mmHg(1 mmHg=0.133 kPa),呼吸18次/分,心率78 次/分,内科查体无异常。神经系统查体:神清,精神可,吐词清晰,双侧瞳孔等大等圆,光反射存在,双眼球活动自如,无眼震及复视,双侧鼻唇沟等称,伸舌居中,双上肢近端肌力Ⅴ级、远端肌力Ⅴ级,双下肢肌力近端肌力Ⅳ+级、远端肌力Ⅴ-级,近端肌力较远端肌力稍差,肌张力正常,腱反射正常,病理征(-),感觉共济粗查可,颈软,脑膜刺激征(-)。
实验室检查:2017年4月23日肌电图提示:双侧胫骨前肌、股四头肌、三角肌可见异常自发活动,运动单位电位(motor unit potential,MUP)时限缩短,多相波增多,且三角肌、股四头肌内侧头波幅降低,呈肌源性损害。肌酶5项:天冬氨酸氨基转移酶99 U/L,乳酸脱氢酶665 U/L,α-羟丁酸脱氢酶829 U/L,肌酸激酶3797 U/L,肌酸激酶同工酶MB 296.57 U/L。血肌炎谱抗体:抗SRP抗体IgG(+++)、抗PM-Scl 75抗体IgG(+)、抗Jo-1抗体IgG(-)、抗Mi-2抗体IgG(-)、抗Ro-52抗体IgG(-)、抗PL-7抗体IgG(-)、抗PL-12抗体IgG(-)、抗EJ抗体IgG(-)、抗OJ抗体IgG(-)、抗PM-Scl 100抗体IgG(-)、抗PM-Scl 175抗体IgG(-),抗核抗体谱均(-)。在本院行左侧股四头肌活检,行组织化学、酶组织化学、免疫组织化学染色。镜下可见,肌纤维直径范围30~130 μm,苏木精-伊红染色和改良Gomori染色可见肌纤维失去正常形态,大小不一,呈圆形化,可见少数小角化纤维,坏死和再生肌纤维。肌间隙增宽,炎性细胞围绕非坏死肌纤维,可见大量结缔组织增生,见图1。肌纤维细胞色素C氧化酶活性正常。腺苷三磷酸环化酶反应,任意视野下均可见两型肌纤维正常分布。酸性磷酸酶活性增加。高碘酸Schiff反应(PAS)提示糖原成分正常。油红O及苏丹黑染色可见肌纤维中脂滴成分正常。免疫组化染色CD4(-),CD8(-),MHC-1(-)。
结合临床特点与病理表现可诊断为抗SRP抗体阳性的多发性肌炎,给予甲强龙500 mg冲击治疗,每3天减半,随后口服甲泼尼龙60 mg/d。14 d后复查肌酶5项:天冬氨酸氨基转移酶56 U/L,乳酸脱氢酶551 U/L,α-羟丁酸脱氢酶671 U/L,肌酸激酶872 U/L,肌酸激酶同工酶MB 180.14 U/L。患者自觉症状较前明显改善,查体未见明显异常,四肢肌力Ⅴ级。
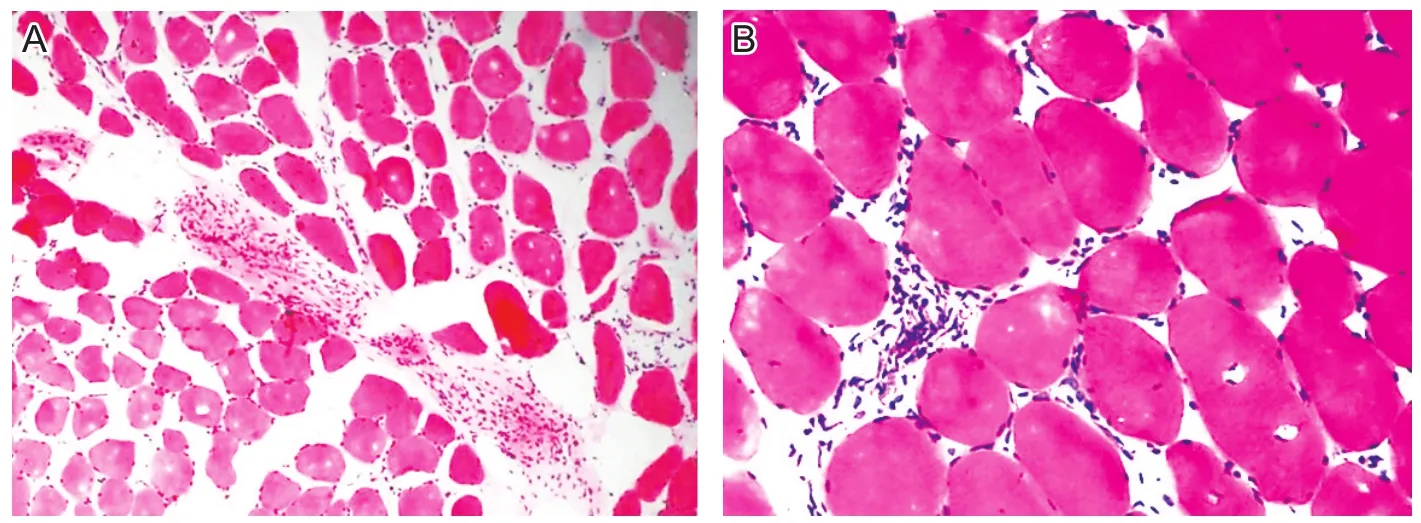
图1 患者左侧股四头肌HE染色结果(光学显微镜)
1月后对患者进行随访,患者目前维持口服甲泼尼龙60 mg/d,症状较前改善,无明显异常,无复发迹象。继续对患者进行随访。
2 讨论
PM的病因和发病机制目前尚不清楚,有学者认为PM与自身免疫紊乱相关,也有研究表明与病毒感染、遗传因素、恶性肿瘤等相关[6]。PM多为亚急性或隐匿起病,多见于18岁以上的成年人,儿童少见,女性发病率大于男性,发病率在各国有所不同,欧美PM最为少见,日本则最为常见,PM在我国发病率不详,但并非最少见[7]。
临床表现上,PM以四肢近端肌肉和颈屈肌受累为主,还可累及肩胛带和骨盆肌。此外,PM可出现疲乏、发热和体重下降等全身症状,可伴有关节痛、关节炎,间质性肺炎、胸膜炎,心律失常、心肌炎,消化道受累,肾脏受累及周围血管受累的雷诺现象等,骨骼肌外受累多见于肌炎特异性抗体阳性的患者[2,6-8]。本例患者为男性,65岁,主要表现为双下肢近端无力,双下肢近端肌力Ⅳ+级、远端肌力Ⅴ-级,伴有双下肢疼痛,起病来体重下降约10 kg,本例患者未发现骨骼肌外受累,余临床特点与PM高度相符。PM患者肌电图常提示存在活动性肌源性损害,肌电图有三联征改变:①时限短、小型的多相运动电位;②纤颤电位、正弦波;③插入性激惹和异常的高频放电。肌电图排除标准包括:①强直性放电提示近端肌强直性营养不良或其他传导通道性病变;②形态分析显示为长时限,大幅多相性运动单位动作电位;③用力收缩所募集的运动单位动作电位类型减少。常规的神经传导检测通常正常,在严重弥漫肌无力患者中可出现复合动作电位波幅降低[7,9-11]。本例患者肌电图提示肌源性损害,符合PM特点。肌肉MRI可以评估骨骼肌的形态、容积和组织结构,显示肌肉的脂肪化、水肿以及代谢改变[12],通过分析的MRI不同的改变有助于鉴别IIM的类型。研究表明,与免疫介导的坏死性肌病相比,多发性肌炎患者骨骼肌水肿比脂肪化明显[13,14]。肌肉MRI已被列为IIM的诊断标准之一,MRI有助于提高肌肉活检的阳性率,并且可以做为判断治疗效果的无创检查手段之一。PM患者肌肉MRI可显示组织内弥漫或片状信号增强,本例患者肌肉MRI提示双侧大腿各组肌群肿胀,异常信号,与PM特点相符。PM患者活动期肌酶五项均升高,其中肌酸激酶最敏感,通常为正常值的5~50倍,血清肌酶不仅具有诊断价值,还与病情轻重、治疗效果及复发情况有关,但肌酸激酶的升高程度并不代表肌无力的严重程度[7,15]。本例患者入院时血清肌酶五项均明显升高,肌酸激酶为3797 U/L,在激素冲击治疗2周后,其水平明显下降,为872 U/L。治疗上,2015年中国多发性肌炎诊治共识[7]推荐使用糖皮质激素、免疫抑制剂及静脉免疫球蛋白。糖皮质激素是治疗PM的首选药物。约1/3的患者治疗后,肌酸激酶及肌酸激酶同工酶MB水平恢复正常,肌力明显改善。本例患者给予激素治疗后,患者自觉症状较前改善,查体未见明显异常,四肢肌力Ⅴ级。有研究表明,糖皮质激素减量或停药后,复发率可达到70%,本例患者曾糖皮质激素冲击治疗3次,均在减量或停药后复发,此次减量1月余,无复发迹象,长期预后需进一步随访。
该患者从临床表现、肌电图结果、肌肉MRI、实验室检查结果以及治疗预后均符合PM特点,以上结果并不能排除免疫介导的坏死性肌病的诊断,其鉴别依赖于肌肉活检。
2004年欧洲神经肌肉疾病中心[1]提出多发性肌炎的病理表现为肌内膜炎细胞(T细胞)围绕并侵入非坏死肌纤维;或肌内膜炎细胞(T细胞)围绕,但不一定侵入非坏死肌纤维;或MHC-1广泛表达。而免疫介导的坏死性肌病的病理表现为肌纤维坏死为主,很少炎性细胞浸润或仅在血管周围少量炎性细胞浸润,肌内膜炎细胞浸润不明显。本例患者肌肉活检病理染色提示:肌纤维直径范围在30~130 μm,失去正常形态,大小不一,呈圆形化,肌间隙增宽,炎性细胞围绕非坏死肌纤维,可见大量结缔组织增生,而未见广泛的肌纤维坏死、再生。这与免疫介导的坏死性肌病具有明显不同,免疫性介导的坏死性肌病病理表现以坏死为主,而PM以炎性细胞浸润为主,该患者符合PM的病理特点。
大多数IIM患者血清中可存在肌炎抗体,Targoff等[16]将肌炎抗体分为3类,包括肌炎特异性抗体、肌炎相关性抗体和组织特异性抗体。1986年,Reeves等[17]在PM患者血清中发现一种针对信号识别颗粒的肌炎特异性抗体,称为抗SRP抗体,在PM中的阳性率为4%~6%[18],而在免疫介导坏死性肌病中阳性率约为16%[19]。抗SRP抗体在多发性肌炎中阳性率明显小于免疫介导的坏死性肌病,有学者将抗SRP抗体作为特异性免疫介导坏死性肌病的标志性抗体[3-5]。值得注意的是,在临床诊疗中,抗SRP抗体阳性常常被认为是免疫坏死性肌病的特异性指标,易被误诊为免疫介导性坏死性肌病。1977年,Wolfe等[20]在PM患者血清中发现抗PM/Scl抗体,抗PM/Scl抗体是一种肌炎相关性抗体,最常见于多发性肌炎/硬化症重叠综合征患者中,阳性率约为25%;也可见于多发性肌炎患者,阳性率约为8%[21]。本例患者抗PM/Scl抗体阳性合并抗SRP抗体阳性,符合PM特点。
综上,本例报道的患者符合多发性肌炎的诊断要点,同时存在抗SPR抗体、抗PM/Scl抗体阳性,因此,二者的鉴别主要依靠肌肉活检,而不能仅依靠肌炎抗体。在肌病的诊断中,不仅要强调血清肌炎抗体的重要性,更要重视肌肉活检的作用。
[1]Hoogendijk JE,Amato AA,Lecky BR,et al.119th ENMC international workshop:trial design in adult idiopathic inflammatory myopathies,with the exception of inclusion body myositis,10-12 October 2003,Naarden,The Netherlands[J].Neuromuscul Disord,2004,14:337-345.
[2]Milisenda JC,Selva-O'Callaghan A,Grau JM.The diagnosis and classification of polymyositis[J].JAutoimmun,2014,48-49:118-121.
[3]Miller T1,Al-Lozi MT,Lopate G,et al.Myopathy with antibodies to the signal recognition particle:clinical and pathological features[J].JNeurol Neurosurg Psychiatry,2002,73:420-428.
[4]Hengstman GJ,Vogels OJ,ter Laak HJ,et al.Myositis during long-term interferon-alpha treatment[J].Neurology,2000,54:2186.
[5]Bronner IM,Hoogendijk JE,Wintzen AR,et al.Necrotising myopathy,an unusual presentation of a steroid-responsive myopathy[J].J Neurol,2003,250:480-485.
[6]Lahouti AH,Christopher-Stine L.Polymyositis and dermatomyositis:novel insights into the pathogenesis and potential therapeutic targets[J].Discov Med,2015,19:463-470.
[7]中华医学会神经病学分会.中国多发性肌炎诊治共识[J].中华神经科杂志,2015,48:946-949.
[8]Lega JC,Fabien N,Reynaud Q,et al.The clinical phenotype associated with myositis-specific and associated autoantibodies:A meta-analysis revisiting the so-called antisynthetasesyndmme[J].Autoimmun Rev,2014,13:883-891.
[9]中华医学会风湿病学分会.多发性肌炎和皮肌炎诊断及治疗指南[J].中华风湿病学杂志,2010,14:828-831.
[10]Bohan A,Peter JB.Polymyositis and dermatomyositis(first of two parts)[J].N Engl J Med,1975,292:344-347.
[11]Bohan A,Peter JB.Polymyositis and dermatomyositis(second of two parts)[J].N Engl J Med,1975,292:403-407.
[12]Theodorou DJ,Theodorou SJ,Kakitsubata Y.Skeletal muscle disease:patterns of MRI appearances[J].Br J Radiol,2012,85:e1298-1308.
[13]Del Grande F,Carrino JA,Del Grande M,et al.Magnetic resonance imaging of inflammatory myopathies[J].Top Magn Reson Imaging,2011,22:39-43.
[14]刘琳琳,杜婧,郝洪军,等.抗信号识别颗粒抗体肌病与多发性肌炎的磁共振成像对比[J].中华神经科杂志,2014,47:232-235.
[15]DimaehkieMM.Idiopathicintlammatory myopathies[J].J Neuroimmunol,2011,231:32-42.
[16]Targoff IN.Update on myositis-specific and myositis-associated autoantibodies[J].Curr Opin Rheumatol,2000,12:475-481.
[17]Reeves WH,Nigam SK,Blobel G.Human autoantibodies reactive with the signal-recognition particle[J].Proc Natl Acad Sci USA,1986,83:9507-9511.
[18]Dimitri D,Andre C,Roucoules J,et al.Myopathy associated with anti-signal recognition peptide antibodies:clinical heterogeneity contrasts with stereotyped histopathology[J].Muscle Nerve,2007,35:389-395.
[19]Christopher-Stine L,Casciola-Rosen LA,Hong G,et al.A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy[J].Arthritis Rheum,2010,62:2757-2766.
[20]Wolfe JF,Adelstein E,Sharp GC.Antinuclear antibody with distinct specificity for polymyositis[J].J Clin Invest,1977,59:176-178.
[21]Brouwer R,Pruijn GJ,van Venrooij WJ.The human exosome:an autoantigenic complex of exoribonucleases in myositis and scleroderma[J].Arthritis Res,2001,3:102-106.
猜你喜欢
现代临床医学(2022年2期)2022-04-19 12:48:52
今日农业(2021年5期)2021-11-27 17:22:19
中国民间疗法(2021年17期)2021-11-04 08:39:30
现代畜牧科技(2021年9期)2021-10-13 06:39:10
现代畜牧科技(2021年5期)2021-07-20 08:07:40
昆明医科大学学报(2021年1期)2021-02-07 01:06:50
反射疗法与康复医学(2017年7期)2017-01-16 01:11:02
兽医导刊(2016年6期)2016-05-17 03:50:50
兽医导刊(2016年6期)2016-05-17 03:50:27
中国当代医药(2015年26期)2015-03-01 02:07:11
