
三氟丙酮酮醇互变异构反应机理的量子化学理论研究
2018-06-27 05:54:58王晓红李宇情李玉莹黄正国
天津师范大学学报(自然科学版) 2018年3期
王晓红 ,李宇情 ,李玉莹 ,黄正国
(1.天津师范大学化学学院,天津300387;2.天津师范大学无机-有机杂化功能材料化学教育部重点实验室,天津300387;3.天津师范大学天津市功能分子结构与性能重点实验室,天津300387)
三氟丙酮(trifluoroacetone)是一种生物活性物质,除了可用于高血压治疗之外,还有多种用途,如可用做防霉剂、除草剂、照相成色剂、兴奋剂、环氧化试剂、有机导体等[1-3].三氟丙酮的衍生物在化工生产中也有重要作用:如一氯三氟丙酮可被用来环合制备含有三氟甲基的精细化学品及咪唑、噻唑、噁唑等医药或农药中间体,生产出的农药具有广谱杂草控制性和优异作物选择性;二溴三氟丙酮与醛基化合物环合形成含有三氟甲基的咪唑类化合物,它们具有很高的抗痛风和抗高尿血酸症活性以及较低的人体副作用[4-7].由于三氟丙酮的酸度比丙酮高且CF3基团具有电负性,所以三氟丙酮在相对温和的碱性条件下可以打断C—C键,成为一种含CF3基团的碳环或杂环的理想构筑基元,从而开辟了多种合成应用的可能性[8].三氟丙酮存在烯醇式(TFP)和酮式(TFA)两种构型,其中的烯醇式构型没有药用价值.如果三氟丙酮的烯醇式构型和酮式构型之间的转化容易进行,就会降低三氟丙酮(酮式构型)的药物活性[3],在一定程度上影响药物生产及应用[9].微波光谱研究表明,气相中三氟丙酮以酮式构型为主,但在碱性溶液中,三氟丙酮的烯醇式构型有可能占据优势地位,可用作合成含氟分子的构筑基元[8].
Favero等[8]报道了三氟丙酮与水分子形成复合物的转动光谱和结构,但水分子在三氟丙酮的烯醇式构型/酮式构型互变异构反应中的作用机制目前尚不清楚.本研究采用量子化学方法,从结构、能量和反应速率等方面综合研究三氟丙酮的烯醇式构型/酮式构型的互变异构反应机理,分析水分子对该酮醇互变异构反应的影响,并预测水溶液中三氟丙酮的烯醇式构型占据优势地位的可能性.
1 计算方法
二阶 Møller-Plessset微扰理论(MP2)是在Hartree-Fock近似的基础上,考虑了电子相关效应后提出的.与Hartree-Fock方法相比,MP2方法在进行弱相互作用体系计算时虽然存在一定的振荡或收敛速度较慢,耗时较长,但计算精度较高[10].本研究运用MP2方法研究三氟丙酮的酮醇互变异构反应机理,并且对所有原子均使用 6-311++G(d,p)基组[11-12]进行优化.首先优化三氟丙酮的烯醇式构型和酮式构型,在此基础上分别构建它们与H2O分子形成的复合物,并在同一计算水平上对所有复合物进行全优化.通过简谐振动频率计算,可以获得零点振动能(ZPVE),并确保获得的复合物具有能量极小值.考虑到基组重叠误差(BSSE)的影响,采用均衡校正法进行BSSE校正[13],以确保复合物和单体均能够在相同的基组下进行优化.采用QST3方法优化无/有水分子参与的酮醇互变异构反应的过渡态构型,进行简谐振动频率的计算,可知所计算的过渡态均具有1个虚频.采用内禀坐标法(IRC)进一步确认过渡态正确地连接了反应物和产物,计算酮醇互变异构反应的活化能,从而进一步计算该反应的速率.另外,使用CPCM溶剂化模型研究溶剂对酮醇互变异构反应的影响.所有计算均采用Gaussian09程序完成.
2 结果与分析
2.1 几何结构
三氟丙酮的酮式构型、烯醇式构型及其与H2O分子形成的复合物的结构如图1所示.
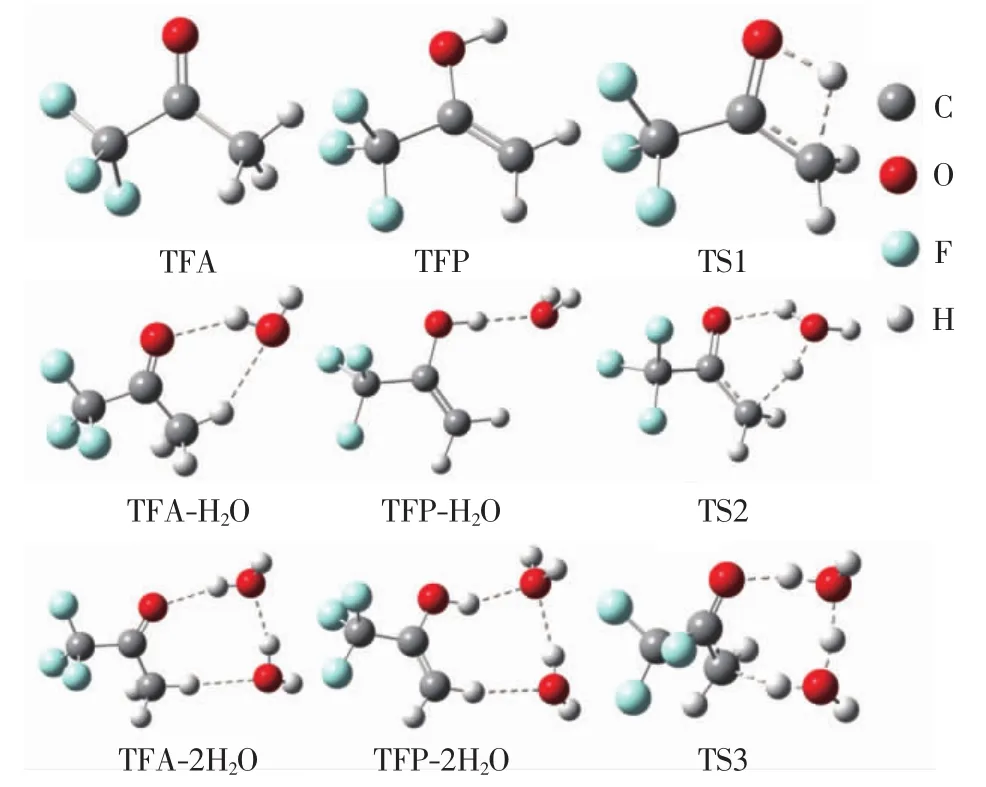
图1 三氟丙酮的烯醇式构型、酮式构型、过渡态及其与H2O分子形成的复合物的结构Fig.1 Scheme of the enol and keto configurations of trifluoroacetone,transition states and their complexes formed with H2O molecules
由图1可知,三氟丙酮酮式构型中的羰基氧原子可作为质子受体形成氢键,而甲基则可作为质子供体形成氢键.烯醇式构型中的羟基和亚甲基可以作为质子供体形成氢键,而其羟基氧原子作为质子受体形成氢键的可能性比较小.尽管烯醇式构型和酮式构型中的F原子都可以作为质子受体形成氢键,但是在这一位置形成氢键的复合物并不是能量最低的稳定构型,且这一氢键与本研究的酮醇互变异构反应无关.此外,无论是在气相还是在水溶液中,三氟丙酮的烯醇式构型、酮式构型、与H2O分子形成的复合物以及过渡态(TS1、TS2、TS3)的结构都相似,这也说明三氟丙酮在水溶液中的酮醇互变异构反应具有与在气相中相似的反应机理.三氟丙酮的酮式构型中甲基上的1个H原子迁移到羰基上,即可形成烯醇式构型,所以该酮醇互变异构反应的过渡态(TS1)中存在1个四元环结构,对应着C—H键的断裂和O—H键的形成.由于四元环结构的张力一般都比较大,所以预测该过渡态的能量比较高.
三氟丙酮的酮式构型与1个H2O分子形成的复合物(TFA-H2O)中有2个分子间氢键,分别为O—HH2O…OTFA和C—HTFA…OH2O,从而构成1个七元环结构,其张力明显小于上述TS1中四元环结构的张力.三氟丙酮的烯醇式构型与1个H2O分子形成的复合物(TFP-H2O)中只有1个O—HTFP…OH2O分子间氢键,所以在TFP-H2O复合物中无环状结构.在1个H2O分子的参与下,三氟丙酮的酮醇互变异构反应是H原子的传递反应,即酮式构型通过把甲基上的1个H原子迁移到H2O分子的O原子上,同时H2O分子的1个H原子迁移到TFA的羰基O原子上,即可生成烯醇式构型,因此该酮醇互变异构反应的过渡态(TS2)中存在1个与TFA-H2O复合物类似的七元环结构.当有1个H2O分子参与反应时,由于TFA-H2O复合物及TS2的七元环结构张力明显小于TS1中四元环结构的张力,因此预测此时三氟丙酮的酮醇互变异构反应会变得较容易进行.
当有2个H2O分子参与反应时,三氟丙酮的酮式构型与2个H2O分子形成的复合物(TFA-2H2O)中有1个八元环结构,它由3个分子间氢键形成,即酮式构型以羰基氧原子和C—H基团为质子受体/供体,分别与一个H2O分子形成分子间氢键O—HH2O…OTFA和C—HTFA…OH2O.此外,2个H2O分子还形成了1个分子间氢键.三氟丙酮的烯醇式构型与2个H2O分子形成的复合物(TFP-2H2O)中也有1个八元环结构,且该八元环结构同样由3个分子间氢键形成.与TFA-2H2O不同的是,TFP-2H2O中2个分子间氢键的质子供体均来自于TFP,分别为TFP的O—H和C—H基团.该酮醇互变异构反应的过渡态(TS3)中存在1个与TFA-2H2O、TFP-2H2O复合物类似的八元环结构.因此,在2个H2O分子的参与下,三氟丙酮的酮醇互变异构反应仍然是1个H原子的传递反应,即酮式构型通过把甲基上的1个H原子迁移到第1个H2O分子的O原子上,同时第1个H2O分子的1个H原子迁移到第2个H2O分子的O原子上,第2个H2O分子的1个H原子迁移到酮式构型的羰基O原子上,从而生成烯醇式构型.同有1个H2O分子参与反应相比,在2个H2O分子参与下的三氟丙酮的酮醇互变异构反应中,H原子传递链变长,从而降低了H原子的迁移效率,不利于烯醇式三氟丙酮的形成.但是,这需要进一步分析H2O分子的参与能否有效降低反应能垒,从而做出综合判断.
理论上,当有更多个H2O分子参与反应时,三氟丙酮的酮醇互变异构反应仍有可能是1个H原子的传递反应,但是随着H原子传递链的增长,H原子的迁移效率可能会降低,从而影响到酮醇互变异构反应的速率.此外,当有更多个H2O分子参与反应时,H2O分子之间的氢键模式变得复杂起来,可能不再形成有利于H原子传递反应的链状氢键,而是形成环状或立体网状氢键,这些环状和立体网状氢键不仅不利于H原子传递反应,而且存在的空间位阻效应也不利于酮醇互变异构反应.本研究通过理论计算表明,当有3个H2O分子参与反应时,三氟丙酮的酮醇互变异构反应的速率较有2个H2O分子参与反应时并无显著变化.所以,仅讨论有1或2个H2O分子参与的情况.
2.2 能量
采用MP2方法优化三氟丙酮烯醇式构型和酮式构型的单体结构,并进一步优化它们与H2O形成的复合物的结构,在同一理论水平上计算复合物的结合能(ΔEb)及烯醇式构型/酮式构型的相对能量差(ΔEr),结果如表1所示.
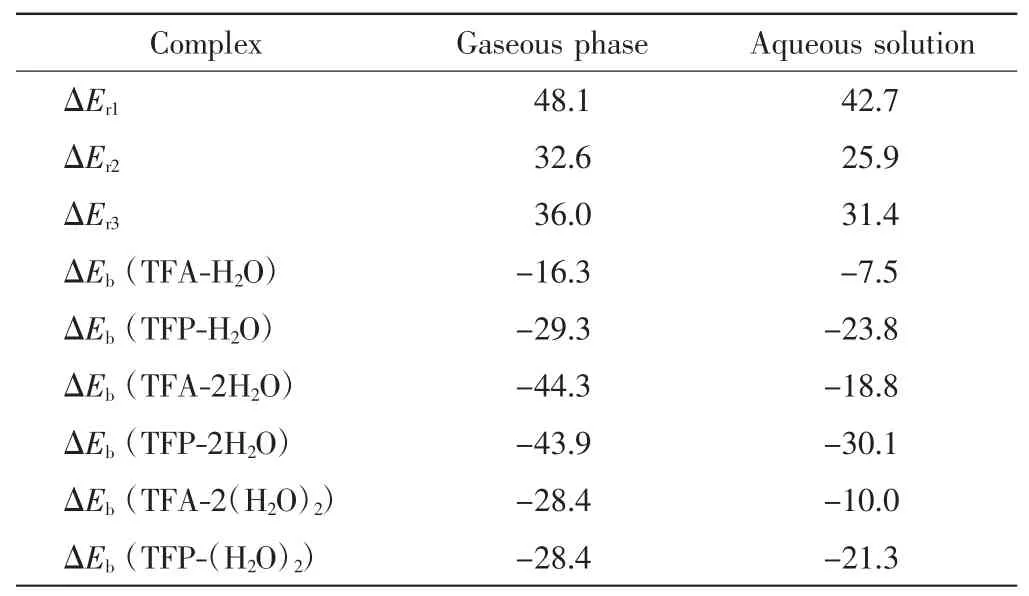
表1 三氟丙酮烯醇式与酮式构型的相对能量差及其与H2O形成复合物的结合能Tab.1 Relative energies between the enol and keto configurations and the binding energies of complexes formed by TFA,TFP interacting with H2O molecules kJ/mol
由表1可知,无论是在气相还是在水溶液中,酮式构型的能量均低于烯醇式构型.因此如果不考虑动力学因素,酮式构型的稳定性高于烯醇式构型,即三氟丙酮主要以酮式构型存在.当有1个H2O分子参与反应时,TFA-H2O与TFP-H2O的相对能量差(ΔEr2)明显减小,可见H2O分子对2种构型的热力学稳定性有显著影响.当有2个H2O分子参与反应时,在气相中TFA-2H2O与TFP-2H2O的相对能量差(ΔEr3)与ΔEr2相比略有增大,可见第2个H2O分子对2种构型热力学稳定性的影响变弱.比较气相与水溶液中的ΔEr,结果发现,水溶液中的ΔEr略小于气相中的ΔEr,这是由溶剂效应造成的,且溶剂效应远小于第1个H2O分子对2种构型热力学稳定性的影响.因此,影响三氟丙酮构型的因素首先是其与H2O分子形成的分子间氢键,其次才是溶剂效应.
在气相中,当有1个H2O分子参与反应时,TFAH2O的结合能为16.3 kJ/mol,明显小于TFP-H2O的结合能(29.3 kJ/mol).这说明尽管TFA-H2O中有2个分子间氢键,但强度比TFP-H2O中的1个分子间氢键弱,也正因如此,才使得TFA-H2O与TFP-H2O之间的相对能量差(ΔEr2)明显减小.当有2个H2O分子参与反应时,如果不考虑2个H2O分子之间的氢键作用,计算出的TFA-2H2O和TFP-2H2O复合物的结合能分别为44.3 kJ/mol和43.9 kJ/mol,比相应的TFA-H2O、TFP-H2O的结合能有显著增加.考虑到2个H2O分子之间的氢键作用,TFA-2H2O和TFP-2H2O复合物的结合能均为28.4 kJ/mol,与TFP-H2O的结合能相当.在水溶液中也有类似的情况.因此当第2个H2O分子参与反应时,复合物的结合能很大一部分来源于2个H2O分子之间氢键作用的贡献.此外,在水溶液中各种复合物的结合能均小于其在气相中的结合能,这是由溶剂效应造成的.
2.3 反应
在上述结构优化的基础上,采用QST3方法在MP2/6-311++G(d,p)水平上计算三氟丙酮由酮式构型向烯醇式构型转化的过渡态,且经IRC计算确认这些过渡态正确连接了反应物和产物.计算由酮式构型→烯醇式构型的活化能,并根据Arrhenius理论计算反应速率,结果如表2所示.

表2 在MP2/6-311++G(d,p)水平上计算的活化能和速率常数Tab.2 Activation energies and rate constants calculated at the MP2/6-311++G(d,p)level
由表2可以看出,没有H2O分子参与时,无论在气相还是水溶液中,三氟丙酮由酮式构型→烯醇式构型的活化能均较高,分别为294.0 kJ/mol和288.6 kJ/mol,因此反应速率非常小,酮式构型占绝对优势,而烯醇式构型几乎可以忽略不计.当有1个H2O分子参与反应时,在气相和水溶液中酮式构型→烯醇式构型的反应活化能显著降低,分别为181.9 kJ/mol和175.2 kJ/mol,导致反应速率常数呈现数量级增加,分别为7.91×10-20s-1和3.76×10-20s-1.当有2个H2O分子参与反应时,酮式构型→烯醇式构型的反应活化能继续降低,但降低程度减弱,在气相和水溶液中活化能分别为144.3 kJ/mol和128.0 kJ/mol,反应速率常数则分别增加到1.52×10-15s-1和6.02×10-11s-1.由此可见,H2O分子的参与有效降低了反应活化能,从而加快了三氟丙酮由酮式构型→烯醇式构型的转化.虽然水溶液中由酮式构型→烯醇式构型转化的活化能比气相中相应的活化能低,但活化能降低的主要因素是H2O分子的参与,溶剂效应是次要因素.尽管TS3的活化能比TS1和TS2都小,但仍然偏高,不会影响酮式构型的主导地位.总之,在中性水溶液中,三氟丙酮的酮式构型虽然可以通过与H2O分子形成氢键复合物,进而转化为烯醇式构型,但由于酮式构型→烯醇式构型转化的反应活化能仍然很高,反应速率较小,因此三氟丙酮在中性水溶液中仍然以酮式构型为主,其药物活性丧失较少.
3 结论
本研究采用MP2方法探讨了三氟丙酮的酮醇互变异构反应机理以及水分子对该酮醇互变异构反应的影响.无论是在气相还是在水溶液中,三氟丙酮的烯醇式构型和酮式构型都可与H2O分子通过分子间氢键形成复合物,这些分子间氢键的形成有效降低了酮醇互变异构反应的活化能,从而使得三氟丙酮更容易由酮式构型转变为烯醇式构型,在此反应中H2O分子起到了催化剂的作用.三氟丙酮的酮醇互变异构反应在水溶液中比在气相中更容易进行,影响酮醇互变异构反应的主要因素是H2O分子的参与,溶剂效应是次要因素.计算三氟丙酮由酮式构型向烯醇式构型转化的活化能,结果发现,中性水溶液中酮醇互变异构反应的活化能仍然很高,不会影响酮式构型的主导地位,所以对三氟丙酮药物活性的影响较小.
[1]SAAIDI P L,GUYONNET M,JEANNEAU E,et al.Trimerization products of trifluoroacetone:Critical solvent effect on position and kinetics of anomeric equilibria[J].J Org Chem,2008,73(4):1209-1216.
[2]MARTIN I,LANGER J,STANO M,et al.Reactions in clusters of acetone and fluorinated acetones triggered by low energy electrons[J].Int J Mass Spectrom,2009,280(1/2/3):107-112.
[3]EVANGELISTI L,FAVERO L B,MARIS A,et al.Rotational spectrum oftrifluoroacetone[J].J Mol Spectrosc,2010,259(2):65-69.
[4]肖恒侨,韩国庆,徐卫国,等.三氟丙酮酸酯的合成与应用研究[J].有机氟工业,2015,46(1):19-23.XIAO H Q,HAN G Q,XU W G,et al.Application and synthesis of 3,3,3-trifluoropyruvate[J].Organo Fluorine Industry,2015,46(1):19-23(in Chinese).
[5]黄晓燕,马正根,林晓霞,等.含苯基吡啶和吩嗪类配体的铱-稀土异金属配合物的合成及作为近红外发光DNA探针[J].科学通报,2014,17(59):1674-1680.HUANG X Y,MA Z G,LIN X X,et al.Synthesis and photophysical properties of iridium-lanthanide complexes containing pyridine and phenazine ligands and NIR probe for DNA[J].Chinese Science Bulletin,2014,17(59):1674-1680(in Chinese).
[6]戴佳亮,徐卫国,金杭丹.1,1-二溴-3,3,3-三氟丙酮的应用进展[J].浙江化工,2015,46(5):1-10.DAI J L,XU W G,JIN H D,Progress in research of 1,1-dibromo-3,3,3-trifluoroacetone[J].Zhejiang Chemical Industry,2015,46(5):1-10(in Chinese).
[7]戴佳亮,徐卫国,金杭丹.1-氯-3,3,3-三氟丙酮的制备与应用[J].有机氟工业,2015,46(3):42-48.DAI J L,XU W G,JIN H D.Progress in preparation and application of 1-chloro-1,1,1-trifluoroacetone[J].Organo Fluorine Industry,2015,46(3):42-48(in Chinese).
[8]FAVERO L B,EVANGELISTI L,MARIS A,et al.How trifluoroacetone interacts with water[J].J Phys Chem A,2011,115(34):9493-9497.
[9]ZEHR R T,DESKINS N A,HENDERSON M A.Photochemistry of 1,1,1-trifluoroacetone on rutile TiO2(110)[J].J Phys Chem C,2010,114(40):16900-16908.
[10]范志辉,陈飞武.二阶多参考态微扰理论计算电子亲和势[J].物理化学学报,2015,31(11):2064-2076.FAN Z H,CHEN F W.Computation of electron affinities with the second order multireference perturbation theory[J].Acta Phys-Chim Sin,2015,31(11):2064-2076(in Chinese).
[11]MCLEAN A D,CHANDLER G S.Contracted gaussian basis sets for molecular calculations.I Second row atoms,Z=11-18[J].J Chem Phys,1980,72(10):5639-5648.
[12]KRISHNAN R,BINKLEY J S,SEEGER R,et al.Self-consistent molecular orbital methods.XX.A basis set for correlated wave functions[J].J Chem Phys,1980,72(1):650-654.
[13]BOYS S F,BERNARDI F.The calculation of small molecular interactions by the differences of separate total energies.Some procedures with reduced errors[J].Mol Phys,1970,19(4):553-566.
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
山东化工(2024年1期)2024-02-04 09:47:12
食品安全导刊(2021年21期)2021-08-30 08:21:52
猪业科学(2018年4期)2018-05-19 02:04:58
合成化学(2015年10期)2016-01-17 08:56:30
分析测试学报(2015年8期)2016-01-13 06:19:33
有机氟工业(2014年3期)2014-06-05 14:36:38
影像科学与光化学(2014年5期)2014-03-11 16:03:04
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
无机化学学报(2014年1期)2014-02-28 17:30:06
