
达托霉素耐药分子机制研究进展
2018-06-26 07:19廖国建彭希希田俊谢建平
生物工程学报 2018年6期
廖国建,彭希希,田俊,谢建平
1 西南大学 药学院 现代生物医药研究所,重庆 400716
2 西南大学 生命科学学院 现代生物医药研究所 三峡库区生态环境与生物资源省部共建国家重点实验室培育基地,重庆 400715
达托霉素 (Daptomycin) 是一种用于治疗革兰氏阳性菌感染的脂肽抗生素,2003年被美国FDA批准用于治疗革兰氏阳性菌引起的复杂皮肤感染和结构性皮肤感染,2006年被批准用于治疗金黄色葡萄球菌导致的菌血症和右侧心内膜炎[1]。2016年我国食品药品监督总局 (CFDA) 批准了华东医药和恒瑞医药生产达托霉素。达托霉素已成为治疗耐甲氧西林金黄色葡萄球菌 (MRSA)和耐万古霉素肠球菌 (VRE) 等高致病性耐药菌导致感染的一线用药。批准上市10余年来致病菌对达托霉素的最小抑菌浓度 (MIC) 没有显著变化[2],耐药比例很低,彰显了达托霉素独特的优势。
由于日益重要的临床价值,达托霉素作用机制和耐药性研究是非常活跃的研究领域。研究主要集中在金黄色葡萄球菌Staphylococcus aureus、枯草芽孢杆菌Bacillus subtilis、肠球菌Enterococcus faecium和Enterococcus faecalis、链球菌Streptococcus mitis和Streptococcus oralis,这些微生物利用不同的适应性途径介导达托霉素耐药(表1)。文中在介绍达托霉素作用机制最新进展的基础上,重点总结了金黄色葡萄球菌和肠球菌耐药基因及其分子机制。
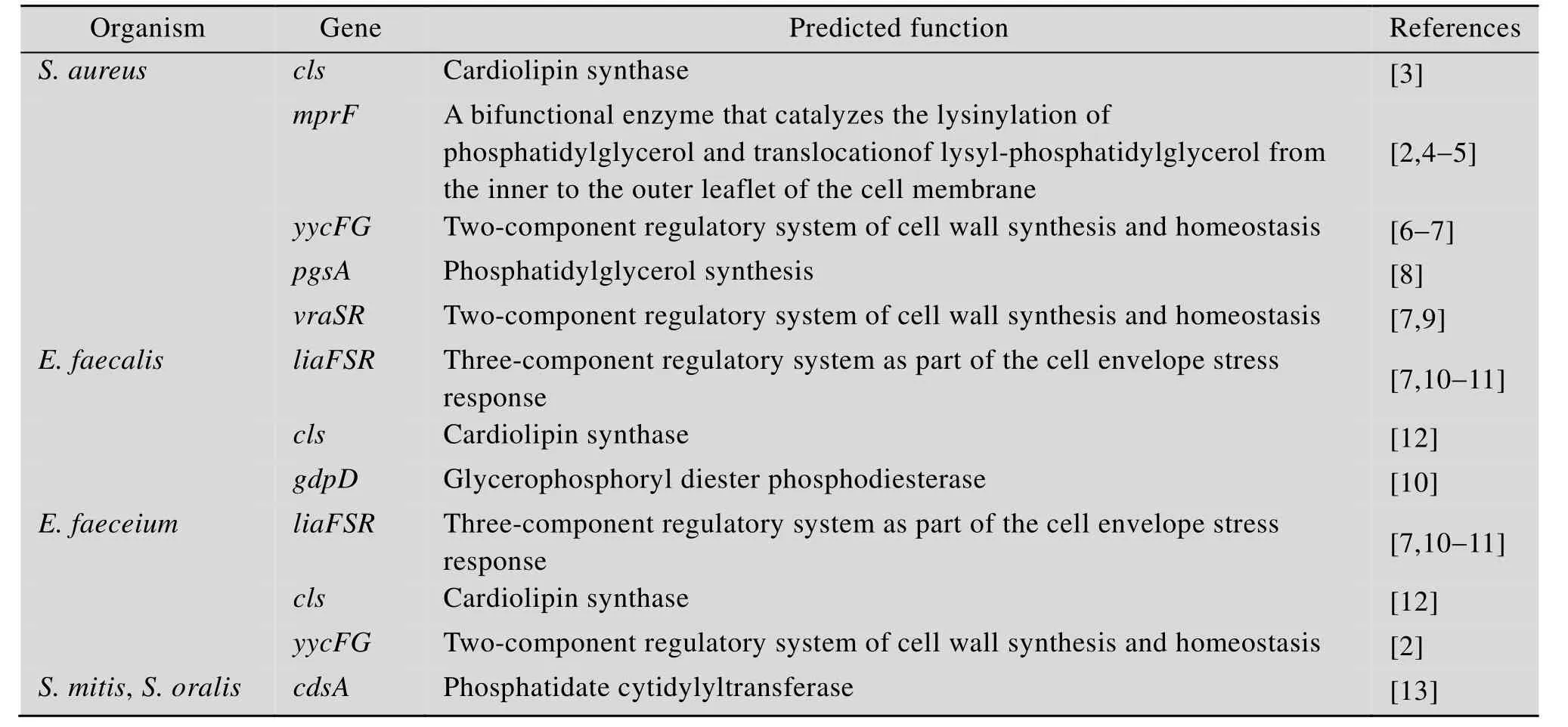
表1 重要致病菌达托霉素耐药相关基因Table 1 Genes associated with daptomycin resistance in major pathogens
1 达托霉素的作用机制
达托霉素通过钙离子 (Ca2+) 依赖的方式靶向革兰氏阳性菌细胞膜[14]。达托霉素与 Ca2+以1∶1的比例结合后寡聚化形成胶束,然后插入到细胞膜中发挥杀菌活性。带正电的达托霉素-钙离子复合物主要与细胞膜中带负电荷的磷脂酰甘油(PG) 和心磷脂 (CL) 等相互作用,导致细胞膜去极化,细胞内容物包括钾离子等的外排,最终导致细胞死亡。达托霉素对细胞壁生物合成的影响和细胞死亡的机制尚未完全阐明。最近的研究表明达托霉素激活了枯草芽孢杆菌细胞膜液脂(Fluid lipid) 微结构域的重排,导致细胞膜逐渐去极化[15]。微结构域重排影响脂质Ⅱ合成酶 MurG和磷脂合成酶PlsX的定位,导致细胞膜磷脂和细胞壁组分合成异常。虽然液脂微结构域也在葡萄球菌和链球菌等病原菌中被观察到,病原菌是否采用类似的机制尚不明确。除了破坏参与细胞壁合成酶的定位外,达托霉素也抑制青霉素结合蛋白Pbp2a的表达。该酶在β-内酰胺抗生素存在的条件下是肽聚糖合成的关键酶。Pbp2a表达量降低恢复MRSA对β-内酰胺抗生素的敏感性。最终β-内酰胺抗生素选择性抑制 Pbp1促进达托霉素结合到细胞膜,从而增加细菌对达托霉素的敏感性,达托霉素抑制Pbp2a的表达导致细菌恢复对β-内酰胺抗生素的敏感性,这种现象称为反向放大效应 (Seesaw effect)[16]。
2 致病菌耐受达托霉素的机制
越来越多的证据表明病原菌主要通过改变细胞壁和细胞膜结构或电荷来耐受达托霉素[7,17]。细胞膜磷脂代谢由多个酶参与 (图 1),合成细胞所需的各种不同磷脂。PG的含量与达托霉素耐药密切相关,降低 PG的合成或增加其转化为赖氨酰磷脂酰甘油 (L-PG) 和 CL的比例都会提高细菌对达托霉素的耐药性。信号转导途径全局性地影响细胞壁的成分和电荷,调控细胞壁的结构动态也是细菌达托霉素耐药的重要因素。
2.1 细胞膜磷脂代谢酶介导达托霉素耐药
2.1.1 MprF
mprF编码一个跨膜双功能酶,由包含14个跨膜结构域的氨基端和1个位于胞质的羧基端所组成。羧基端负责催化磷脂酰甘油 (PG) 的赖氨酰化,氨基端跨膜结构域具有翻转酶活性,负责将 L-PG从细胞膜内侧转位到细胞膜外侧[18]。由于 L-PG带正电荷的性质,导致细胞膜外侧负电荷减少。
mprF突变与金黄色葡萄球菌达托霉素耐药相关,mprF缺失导致细菌对达托霉素更敏感[19]。此外,通过反义RNA技术特异性地降低mprF的转录也导致耐药菌株恢复对达托霉素的敏感性[20]。在缺失mprF的突变株中,表达耐药突变株中的mprF导致回补菌株提高了达托霉素抗性,而表达野生型菌株的mprF则没有这种效果。分析耐药菌株的mprF序列发现,mprF突变主要集中在酶的热点区域 (图 2),主要位于中央跨膜结构域和羧基端结构域,这些突变很多是功能获得性(Gain-of-function) 突变,能够增加mprF的表达量或者提高酶的活性[2]。肠球菌中含有两个mprF,其中mprF2发挥主要作用,其具有更广泛的底物范围,能够形成赖氨酸、丙氨酸和精氨酸修饰的 PG。敲除mprF2导致对多粘菌素等抗菌肽更敏感,然而对达托霉素的抗性没有改变,这表明达托霉素耐药机制可能具有菌株特异性。

图1 革兰氏阳性细菌主要磷脂结构及其生物合成途径Fig. 1 Chemical structure (A) and biosynthetic pathway(B) of the major phospholipids in Gram-positive bacteria.PA: phosphatidic acid; CDP-DAG: CDP-diacylglycerol;PS: phosphatidylserine; PE: phosphatidylethanolamine;G-3-P: glycerol-3-phosphate; PG-P: phosphatidylglycerol-3-P; PG: phosphatidylglycerol; CL: cardiolipin.

图2 达托霉素耐药金黄色葡萄球菌中发现的 MprF突变位点Fig. 2 Mutations in MprF identified in daptomycin resistant S. aureus. TM: transmembrane domain.
研究发现并非所有的mprF突变都改变了细胞膜的电荷[11]。此外,人工磷脂囊泡中改变L-PG的含量对达托霉素的结合仅有很小的影响[12],因此MprF可能通过控制PG的浓度、改变细胞膜脂类的分布等因素影响达托霉素的寡聚化和介导达托霉素耐药。
2.1.2 Cls
cls编码心磷脂合成酶,利用两分子PG合成一分子CL,负责细胞膜心磷脂的生物合成。该酶的突变会改变细胞膜中PG和CL的比例。在金黄色葡萄球菌中,Cls 的K61缺失和位于磷脂酶D结构域的R218Q突变等参与达托霉素耐药[3]。在敏感菌株中过表达含 R218Q突变基因显著提高达托霉素抗性,导致菌株的MIC从4 μg/mL上升为64 μg/mL。同样的R218Q突变也在耐药屎肠球菌被发现,这些结果暗示Cls也是肠球菌中达托霉素耐药的关键酶[12]。异源表达纯化R218Q突变蛋白和酶活性测定发现,突变蛋白合成心磷脂的能力增加,令人意外的是,突变株和野生型菌株相比心磷脂含量并未发生显著变化。二维薄层色谱分析细胞膜磷脂时发现,耐药菌株比野生型菌株中含有更低的 PG和甘油磷酸-二酰甘油含量 (CDP-DAG)[21]。这表明增强 Cls活性可能通过降低细胞膜中的PG含量进而改变细胞膜的特征。这种效果与金黄色葡萄球菌中mprF突变效果类似。
2.1.3 CdsA
缓症链球菌和口腔链球菌容易产生高度达托霉素耐药突变株 (>256 μg/mL)。测序3株耐药菌时发现,突变株都含有cdsA基因突变 (Q31*、G246C和 D249N)(13)。cdsA编码胞苷酰转移酶负责催化磷脂酸 (PA) 为CDP-DAG。cdsA在多数细菌中都是必需基因,合成细胞生存所需的各种脂类。令人意外的是,由于尚不清楚的机制,cdsA在缓症链球菌和口腔链球菌中是非必需基因。cdsA突变导致菌株完全丧失了合成PG和CL的能力,大量积累PA。筛选这3株耐药菌株回复突变株时发现,Q31*的终止密码子转变为色氨酸密码子,使得蛋白能够正常翻译;C246突变为丝氨酸密码子,恢复该区域的柔性;N249则恢复为野生型天冬氨酸密码子。这些结果表明cdsA介导缓症链球菌和口腔链球菌达托霉素耐药性。

图3 达托霉素耐药肠球菌 (蓝色) 和金黄色葡萄球菌 (红色) 中Cls突变位点[2]Fig. 3 Mutations in Cls identified in daptomycin resistant S. aureus (red) and Enterococci (blue)[2]). TM:transmembrane domain; PLD: phospholipase domain.
2.1.4 PgsA
细菌可以调控细胞膜磷脂合成的不同步骤来影响PG的含量。除了前文提到的cdsA、cls和mprF外,还包括pgsA等。pgsA编码磷脂酰甘油合成酶,催化CDP-DAG合成磷脂酰甘油。枯草芽孢杆菌中,达托霉素耐药菌株中含有pgsA突变 (A64V)[8],耐药菌株中PG的含量比野生型低5倍,达托霉素MIC提高30倍。
2.2 细胞外膜应激调控网络
除了细胞膜磷脂代谢关键酶外,细胞外膜应激调控网络介导的细胞全局性改变也在达托霉素耐药过程中发挥了重要作用。在金黄色葡萄球菌中主要涉及双组分系统VraSR和YycFG,而在肠球菌中主要涉及三组分系统LiaFSR。
2.2.1 YycFG (WalKR)
YycFG (WalKR) 是金黄色葡萄球菌等低G+C革兰氏阳性致病菌中的重要双组分系统,调控细胞壁合成和代谢、细胞膜完整性、细胞分裂、脂类代谢等多种功能[22]。YycF为应答调节蛋白,YycG为组氨酸蛋白激酶。YycG的胞内部分可分为HAMP连接子、PAS折叠感应域、二聚化组氨酸磷酸转移域和ATPase酶结构域。这个系统还包括辅助基因yycI和yycH。YycH和YycI都是氨基端跨膜蛋白,胞外的羧基端结构域在枯草芽孢杆菌中抑制YycG 组氨酸激酶活性[15]。在非分裂的细胞中,YycFGHI复合体在整个细胞膜中都有分布,在细胞分裂时 YycG定位于细胞分裂处,该位点也是达托霉素在细胞膜上的插入位点。YYcG可能通过感受细胞膜流动性的改变,进而调整细胞壁的交联度来应对细胞应激。
在耐药菌株中发现了数个YycG的突变位点,如位于PAS (R236C) 和HAMP (S221P) 的突变。另一个研究发现效应蛋白WalkR的突变也与达托霉素耐药相关,A96T突变位于保守的区域,影响磷酸化介导的蛋白构象改变;而K208R突变位于DNA结合区域,突变导致MIC提高4倍[6]。耐药突变也发生在辅助基因有yycH,移码突变导致蛋白截短。yycG突变也在耐药粪肠球菌中被观察到。达托霉素耐药表型与yycFG表达不足的表型类似,突变可能影响了操纵子的功能,下调细胞壁结构动态来应对达托霉素的攻击。
2.2.2 VraSR
VraSR 是低 G+C革兰氏阳性细菌中高度保守的双组分系统,通过调控pbp2(青霉素结合蛋白 2)、tagA(参与 WTA 合成)、prsA(伴侣蛋白) 和murA(UDP-N-乙酰葡萄糖胺烯醇式丙酮酸转移酶) 的转录控制细胞壁的合成[7]。达托霉素处理导致金黄色葡萄球菌vraSR表达上调。在耐药菌株中失活vraSR导致菌株达托霉素敏感,并伴随更薄的细胞壁。上调vraSR的转录与达托霉素耐药相关。此外,vraS的点突变 (E276K) 也导致耐药[9]。
2.2.3 LiaFSR
肠球菌利用 LiaFSR三组分调控系统应对细胞膜压力,类似于金黄色葡萄球菌的VraSR调控系统。在枯草芽孢杆菌中,LiaFSR系统在达托霉素存在条件下被激活。liaFSR基因突变在耐药肠球菌中被发现。在耐药粪肠球菌中发现了liaF的突变,突变导致Ile177缺失。在敏感菌株中替换耐药株的liaF导致MIC提高4倍[10]。许多耐药屎肠球菌菌株含有liaFSR突变[7]。liaR缺失导致心磷脂的重新分配和达托霉素远离分裂位点[11]。
2.3 其他机制
最近的研究表明,金黄色葡萄球菌通过主动释放细胞膜磷脂作为诱饵来结合和失活达托霉素,阻止达托霉素结合到细胞膜上[23]。该过程受Agr密度感应系统的调控,细菌通过表达一种小分子肽毒素 PSMα削弱这种防护机制。苯唑西林的使用也能抑制细胞膜磷脂的释放。达托霉素的高度耐药常常需要多个基因的参与,粪肠球菌达托霉素耐药菌株中发现了gdpD(I170 缺失) 和liaF(I177缺失) 突变。gdpD编码的跨膜蛋白参与磷脂代谢。野生型菌株中导入突变株的liaF导致突变菌株的MIC从1 μg/mL变为4 μg/mL,单独导入gdpD不足以获得达托霉素抗性。有趣的是,当野生型菌株同时引入突变的liaF和gdpD时,达托霉素的耐药性变为12 μg/mL,这表明这两个基因相互作用,导致了高水平达托霉素耐药性[10]。
2.4 达托霉素耐药的工作模型
归纳整合已有证据构建病原菌耐药的工作模型如图4所示。重要致病菌主要通过改变细胞膜磷脂代谢关键酶的表达或者活性,控制细胞膜中PG的含量,以及直接释放PG到细胞外捕获达托霉素-钙离子复合物,最终降低达托霉素与细胞膜的结合量。此外,通过信号转导途径全局性地影响细胞壁的成分和电荷,调控细胞壁的稳态,降低达托霉素通过细胞壁的能力。细菌整合多种耐药机制最终实现达托霉素耐药。

图4 达托霉素耐药机理Fig. 4 Resistance to daptomycin. The membrane-associated proteins involved in phospholipid metabolism(CdsA, PgsA, MprF and Cls) play an important role in daptomycin resistance through controlling the content and concentration of PG. Furthermore, daptomycin can be sequestered by the free PG and can be repelled by the structure or charge change of important cell wall ingredients. The cell wall homeostasis is under control of important signal transduction pathway, such as YycFG,VraSR and LiaFSR.
3 总结与展望
过去 10年,多重耐药革兰氏阳性菌株的显著增加使达托霉素成为治疗严重感染的重要选择。达托霉素强力杀菌活性和独特的杀菌机理使其受到临床医生的青睐。达托霉素的大量使用,使得耐药菌株也变得越来越普遍。此外,放线菌如达托霉素产生菌玫瑰孢链霉菌和结核分支杆菌对达托霉素天然耐药,限制了这种重要抗生素用于防控日益严重的结核病威胁,笔者课题组正在利用功能基因组学手段揭示结核菌和玫瑰孢链霉菌达托霉素耐药的分子机制,研究结果有望丰富耐药的途径和方式,并为达托霉素用于结核病治疗奠定基础。随着耐药菌株耐药分子机制的进一步阐明和宿主环境对达托霉素敏感性影响机理的深入研究,将为提高达托霉素活性的新治疗手段提供重要靶点,为感染性疾病的防控发挥更大的作用。
REFERENCES
[1]Lee SY, Fan HW, Kuti JL, et al. Update on daptomycin:the first approved lipopeptide antibiotic. Expert Opin Pharmacother, 2006, 7(10): 1381–1397.
[2]Humphries RM, Pollett S, Sakoulas G. A current perspective on daptomycin for the clinical microbiologist. Clin Microbiol Rev, 2013, 26(4):759–780.
[3]Peleg AY, Miyakis S, Ward DV, et al. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates ofStaphylococcus aureus. PLoS ONE, 2012, 7(1): e28316.
[4]Ernst CM, Staubitz P, Mishra NN, et al. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog, 2009, 5(11): e1000660.
[5]Rubio A, Conrad M, Haselbeck RJ, et al. Regulation ofmprFby antisense RNA restores daptomycin susceptibility to daptomycin-resistant isolates ofStaphylococcus aureus. Antimicrob Agents Chemother,2011, 55(1): 364–367.
[6]Howden BP, McEvoy CR, Allen DL, et al. Evolution of multidrug resistance duringStaphylococcus aureusinfection involves mutation of the essential two component regulator WalKR. PLoS Pathog, 2011, 7(11):e1002359.
[7]Miller WR, Bayer AS, Arias CA. Mechanism of action and resistance to daptomycin inStaphylococcus aureusandEnterococci. Cold Spring Harb Perspect Med, 2016,6(11): a026997.
[8]Hachmann AB, Angert ER, Helmann JD. Genetic analysis of factors affecting susceptibility ofBacillus subtilisto daptomycin. Antimicrob Agents Chemother,2009, 53(4): 1598–1609.
[9]Su J, Iehara M, Yasukawa J, et al. A novel mutation in thevraSgene ofStaphylococcus aureuscontributes to reduce susceptibility against daptomycin. J Antibiot(Tokyo), 2015, 68(10): 646–648.
[10]Arias CA, Panesso D, McGrath DM, et al. Genetic basis forin vivodaptomycin resistance inEnterococci. New Engl J Med, 2011, 365(10): 892–900.
[11]Reyes J, Panesso D, Tran TT, et al. AliaRdeletion restores susceptibility to daptomycin and antimicrobial peptides in multidrug-resistantEnterococcus faecalis. J Infect Dis, 2015, 211(8): 1317–1325.
[12]Avlieva M, Zhang WN, Arias CA, et al. Biochemical characterization of cardiolipin synthase mutations associated with daptomycin resistance inEnterococci.Antimicrob Agents Chemother, 2013, 57(1): 289–296.
[13]Mishra NN, Tran TT, Seepersaud R, et al. Perturbations of phosphatidate cytidylyltransferase (CdsA) mediate daptomycin resistance inStreptococcus mitis/oralisby a novel mechanism. Antimicrob Agents Chemother, 2017,61(4): e02552–16.
[14]Baltz RH. Daptomycin: mechanisms of action and resistance, and biosynthetic engineering. Curr Opin Chem Biol, 2009, 13(2): 144–151.
[15]Müller A, Wenzel M, Strahl H, et al. Daptomycin inhibits cell envelope synthesis by interfering with fluid membrane microdomains. Proc Natl Acad Sci USA,2016, 113(45): E7077–E7086.
[16]Renzoni A, Kelley WL, Rosato RR, et al. Molecular bases determining daptomycin resistance-mediated resensitization to β-lactams (Seesaw Effect) in methicillin-resistantStaphylococcus aureus. Antimicrob Agents Chemother, 2017, 61(1): e01634–16.
[17]Tran TT, Munita JM, Arias CA. Mechanisms of drug resistance: daptomycin resistance. Ann New York Acad Sci, 2015, 1354(1): 32–53.
[18]Ernst CM, Peschel A. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol Microbiol, 2011,80(2): 290–299.
[19]Ernst CM, Staubitz P, Mishra NN, et al. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog, 2009, 5(11): e1000660.
[20]Rubio A, Conrad M, Haselbeck RJ, et al. Regulation ofmprFby antisense RNA restores daptomycin susceptibility to daptomycin-resistant isolates ofStaphylococcus aureus. Antimicrob Agents Chemother,2011, 55(1): 364–367.
[21]Mishra NN, Bayer AS, Tran TT, et al. Daptomycin resistance inEnterococciis associated with distinct alterations of cell membrane phospholipid content. PLoS ONE, 2012, 7(8): e43958.
[22]Bayer AS, Schneider T, Sahl HG. Mechanisms of daptomycin resistance inStaphylococcus aureus: role of the cell membrane and cell wall. Ann New York Acad Sci, 2013, 1277(1): 139–158.
[23]Pader V, Hakim S, Painter KL, et al.Staphylococcus aureusinactivates daptomycin by releasing membrane phospholipids. Nat Microbiol, 2016, 2: 16194.
猜你喜欢
心肺血管病杂志(2020年5期)2021-01-14
三农资讯半月报(2020年18期)2020-10-14
中成药(2019年12期)2020-01-04
数码世界(2018年1期)2018-12-23
中成药(2018年7期)2018-08-04
浙江工业大学学报(2017年5期)2018-01-22
中成药(2017年12期)2018-01-19
中成药(2017年5期)2017-06-13
饮食科学(2016年9期)2016-11-18
中国医疗美容(2015年4期)2015-04-27
