
UPLC-MS/MS法检测唾液中的奥沙西泮
2018-06-25 08:52吴东星李树华李继印任国印曾思火
食管疾病 2018年2期
吴东星,李树华,李继印,任国印,曾思火,李 虹
随着滥用药物逐渐成为世界性的社会问题,体内滥用药物分析向法庭科学领域、毒物分析领域提出了新的任务和要求。目前,国内对滥用药物检测的方法有:气相色谱—质谱联用(gas chromatography/mass spectrometry,GC/MS)法[2-4]、高效液相色谱(high performance liquid chromatography,HPLC)法[5-8]和超高效液相色谱—串联质谱(ultra performance liquid chromatography-mass spectrometry/mass spectrometry,UPLC-MS/MS)法[9-13],多用以检测人体血液、尿液,而对唾液检测的文献报道较少。因此,本研究用UPLC-MS/MS法对人体唾液中奥沙西泮(oxazepam)进行了检测,现报道如下。
1 材料与方法
1.1仪器超高效液相色谱—串联质谱联用仪:AgiLent1290型超高效液相色谱仪(安捷伦科技有限公司,德国)和AB SCIEX 4000Q TRAP 串联质谱仪(AB SCIEX公司,新加坡),配Analyst® software工作站;AllegraTM X-22台式高速离心机(贝克曼库尔特公司,美国)。
1.2标准品与试剂奥沙西泮购自公安部物证鉴定中心,纯度≥98%;乙腈、甲醇试剂为色谱纯,甲酸、氨水试剂为分析纯,实验用水为去离子水。
1.3实验样品65份唾液样品均采自口服奥沙西泮1片(每片15 mg)后30 min 至12 h的5名志愿者。
1.4色谱条件ACQUITY UPLC® BEH C18色谱柱(2.1 mm×50 mm,1.7 μm);流动相0.1%甲酸水溶液—乙腈梯度洗脱,洗脱程序见图1;进样体积5 μL,流速0.4 mL·min-1,运行时间6 min,柱温35 ℃。
1.5质谱条件采用电喷雾离子源(electron spray ionization,ESI),分析物在正离子扫描下检测;三重四极杆检测器;毛细管电压为5.5 kV;去溶剂气温度550 ℃,去溶剂气流量:800 L·h-1;锥孔气流量30 L·h-1;碰撞气流量:0.15 mL·min-1,锥孔电压80/12,碰撞电压30/13。奥沙西泮的监测离子为287.0/241.0、287.0/165.0和287.0/104.0。
1.6样品前处理用甲醇稀释唾液至一定倍数,经振荡器振荡3 min,在15 000 r·min-1转速下离心3 min,取上清液过有机滤膜(13 mm×0.22 μm)后供检。
1.7标准工作液的配制精密称量10.0 mg奥沙西泮标准品于10 mL容量瓶内,用甲醇稀释至刻度线,振摇均匀得到1.0 mg·mL-1奥沙西泮标准液,置4 ℃冰箱保存。取空白唾液,按1.6节方法处理后,取适量上清液按浓度梯度逐次稀释1.0 mg·mL-1奥沙西泮标准液成浓度分别为100、50、20、10、5和1 ng·mL-1的线性工作溶液,置4 ℃冰箱保存、待测。
2 结果
2.1系统适应性试验在1.4和1.5分析条件下,分别对空白对照、质控样品(10 ng·mL-1)和唾液样品检测,以287.0/241.0、287.0/165.0和287.0/104.0这3对离子对和保留时间(Rt=2.24 min)进行定性分析,并与已知奥沙西泮标准物质进行比对,空白对照无干扰。进样结果表明:UPLC-MS/MS检测方法专属性好,见图1。
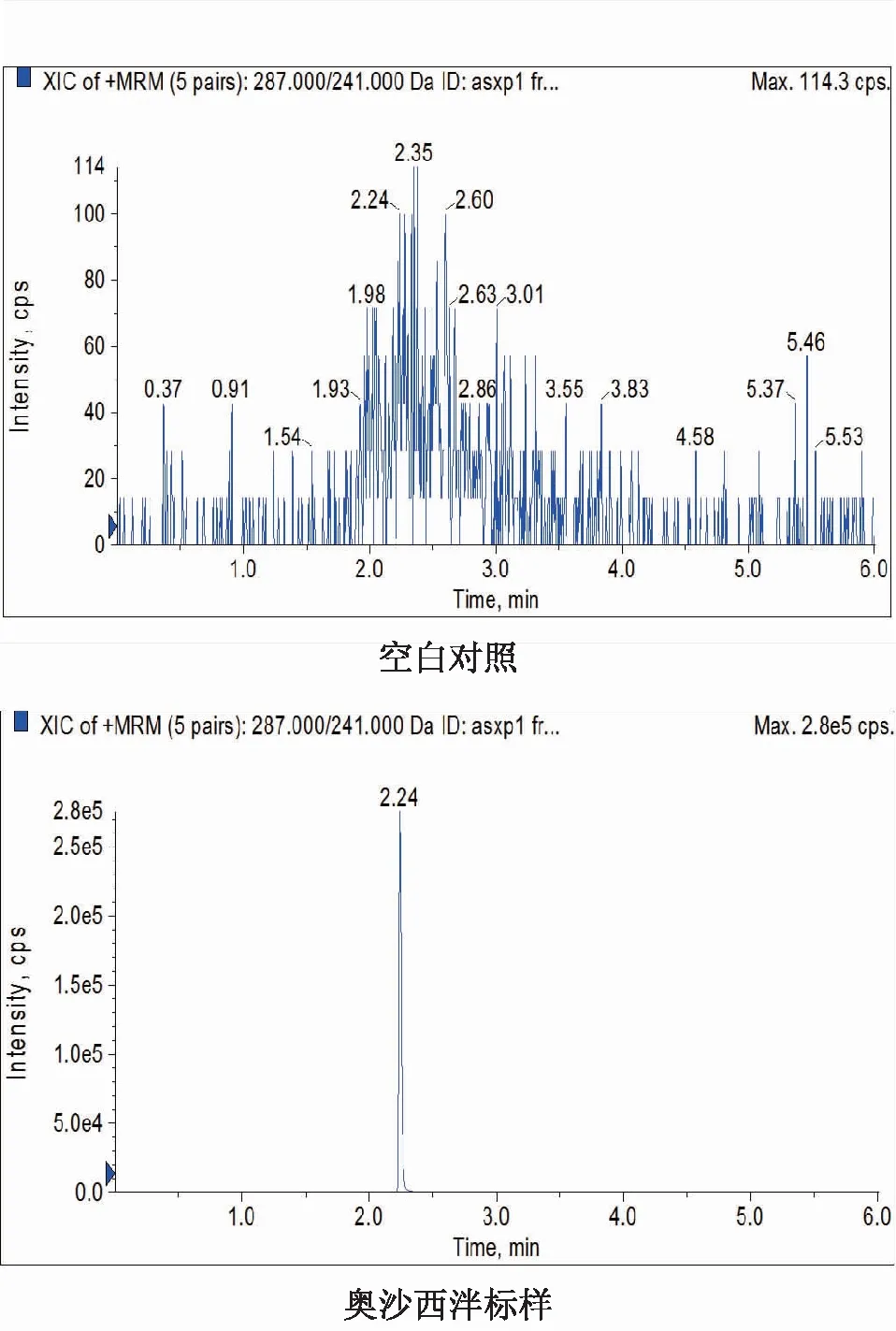
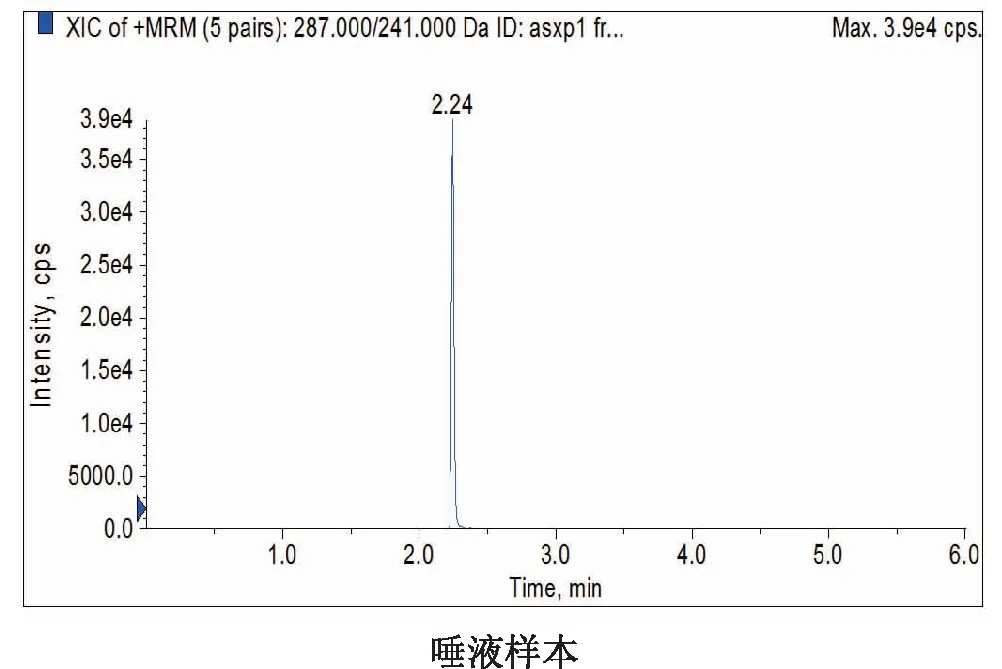
图1 不同样品中奥沙西泮的定量离子对色谱图
2.2流动相的考察本文分别用流动相为0.1%甲酸的乙腈溶液-0.1%氨水溶液和乙腈-0.1%甲酸水溶液对质控样品进行梯度洗脱,进样结果表明:奥沙西泮在流动相为乙腈-0.1%甲酸水溶液中响应度较高。因此,本文选用乙腈-0.1%甲酸水溶液作为流动相。
2.3提取溶剂的考察本文分别用乙腈、甲醇试剂对唾液样品进行前处理,经UPLC-MS/MS检测分析表明:用甲醇作为提取溶剂时,奥沙西泮具有良好的色谱峰。
2.4线性范围、检出限和定量限取浓度分别为1、5、10、20、50和100 ng·mL-1的奥沙西泮标准溶液,在1.4和1.5节条件下测定;以定量离子(287.0/241.0)峰面积A为纵坐标,质量浓度C为横坐标,绘制奥沙西泮的标准曲线,求得标准曲线的回归方程和相关系数。以线性最低点溶液逐倍稀释后,以信噪比>3为检出限,以信噪比>10为定量限。奥沙西泮的线性方程、检出限和定量限见表1。
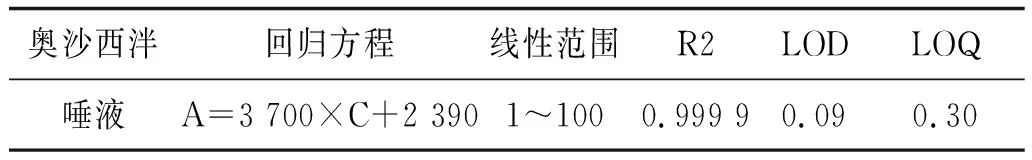
表1 奥沙西泮的线性方程、检出限和定量限 ng·mL-1
2.5回收率、基质效应和精密度分别配制5、50、90 ng·mL-1奥沙西泮标准溶液各6份,按1.6节方法处理后,在1.4和1.5节条件下测定,每日进样5次,每次间隔2 h,记录峰面积As,计算日内精密度(relative standard deviation,RSD)表示;每份样分别连续测定3 d,计算日间精密度用相对标准偏差。取空白唾液18份,分别加入浓度为5、50、90 ng·mL-1的奥沙西泮标准溶液1 mL,配置成实际添加含量分别5、50、90 ng的空白添加系列样本为各6份,按1.6节方法处理后,在1.4和1.5节条件下测定,记录峰面积为Am。将所测得的Am数据代入奥沙西泮标准曲线方程,并计算测得值,以测得值与实际添加量对比,计算提取回收率。另取上述3个浓度的标准溶液,在1.4和1.5节条件下直接进样测定,记录峰面积为Astd,以Am/Astd计算基质效应,结果见表2。
表2 奥沙西泮的回收率、基质效应、准确度、精密度和稳定性
注:RSD:相对标准偏差。
2.6稳定性配制低、中、高3个浓度的质控样品(n=3),于室温放置24 h及经过3次冻融循环后,按1.6节方法处理质控样品浓度为5、50、90 ng·mL-1的溶液,并在1.4和1.5节条件下测定,考察其短期稳定性及冻融稳定性。结果表明,奥沙西泮在上述条件下均稳定(见表4)。
2.7样品测定采集5位志愿者的65份唾液,按1.4节方法处理后,在1.4和1.5节条件下测定。65份唾液中,全部检出奥沙西泮,其中定量数为57份,8份只能定性,结果见表3。
3 讨论
本研究建立了唾液中奥沙西泮的UPLC-MS/MS的检测方法。实验结果表明:唾液样品经甲醇蛋白沉淀后,以流动相为乙腈-0.1%甲酸水溶液对奥沙西泮进行梯度洗脱,以电喷雾离子源正离子多反应监测模式进行质谱分析,并通过多个特征离子对和保留时间对唾液中奥沙西泮进行定性分析。用UPLC-MS/MS法检测唾液中的奥沙西泮,该检测方法提取回收率高,大于90%;基质效应低,唾液样品无需加酶水解,样品前处理简便;检测方法灵敏度高,检出限低于0.1 ng·mL-1,准确度高。
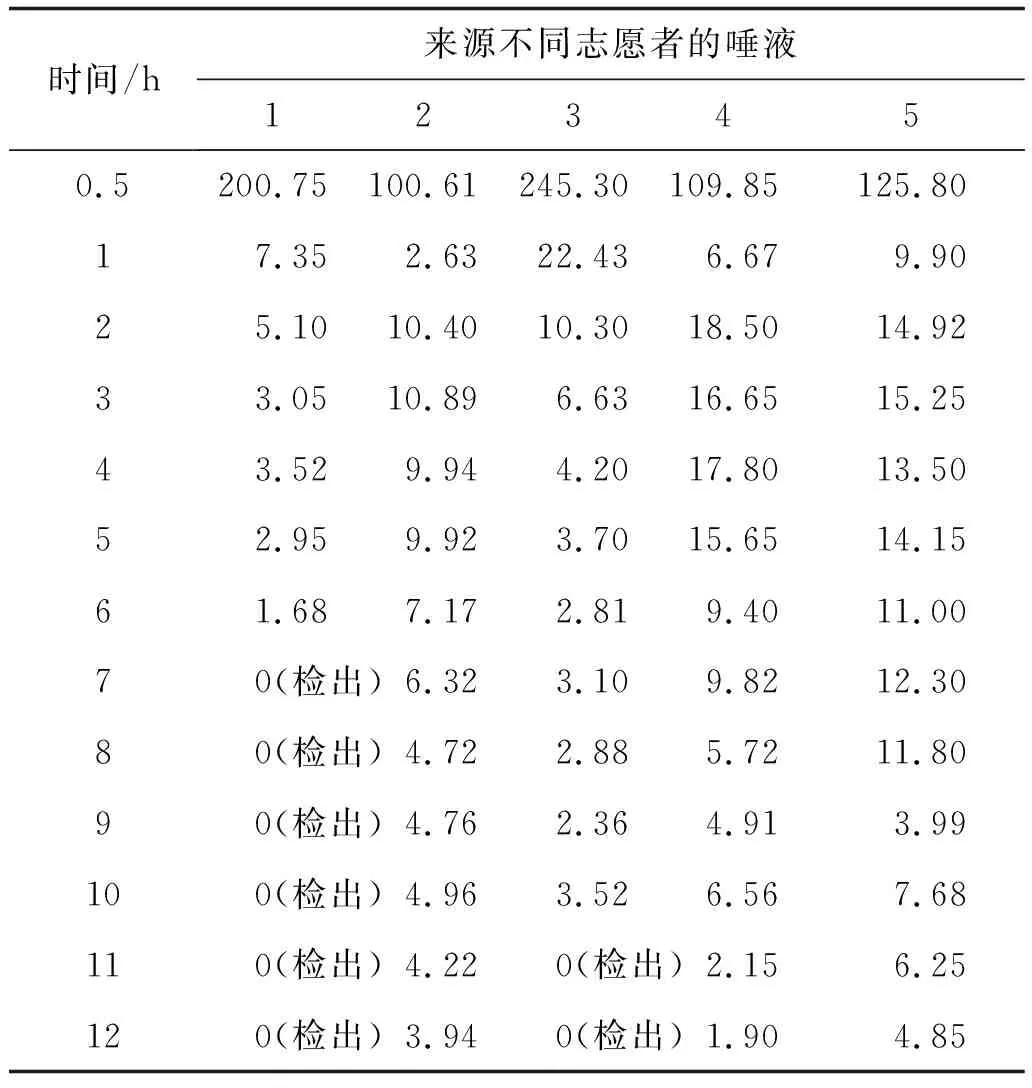
表3 唾液样品中奥沙西泮的检测结果 ng·mL-1
5名志愿者65份唾液样品中均检出奥沙西泮成分,其中57份唾液样品中奥沙西泮的含量高于定量限,8份仅能定性分析,研究表明:在人服用奥沙西泮12 h内,唾液中仍可检出一定量的奥沙西泮,且含量相对稳定。因此,可以选取唾液样品作为生物样品中奥沙西泮的检材,为体内滥用精神活性药物检测提供了一个有效的分析方法。
参考文献:
[1] 罗涛,郝伟.奥沙西泮的研究进展[J].国际精神病学杂志,2010,37(4):244-247.
[2]Simonsen KW,Hermansson S,Steentoft A,et al.A validated method for simultaneous screening and quantification of twenty-three benzodiazepines and metabolites plus zopiclone and zaleplone in whole blood by liquid-liquid extraction and ultra-performance liquid chromatography-tandem mass spectrometry[J].J Anal Toxicol,2010,34(6):332-341.
[3]Goldberger BA,Chronister CW,Merves ML.Quantitation of benzodiazepines in blood and urine using gas chromatography-mass spectrometry(GC-MS)[J].Methods Mol Biol,2010,603:75-87.
[4] 朱小红,李涛,李建荣.苯二氮类药物的气相色谱—质谱联用法定性分析方法研究[J].药物分析杂志,2011,31(12):2289-2293.
[5] 张韬,凌义.正相液相色谱法测定人体血浆中奥沙西泮的浓度[J].中国药物警戒,2012,9(5):257-259.
[6] 郑付春,江泽斌.固相萃取—液相色谱法测定血浆中4种苯二氮类药物[J].汕头大学医学院学报,2014,27(4):208-210.
[7] 徐晖.高液相色谱法同时测定苯巴比妥、卡马西平、艾司唑仑、阿普唑仑、氯硝西泮、奥沙西泮的血药浓度[J].海峡药学,2013,25(9):170-173.
[8] 刘立新,崔一民.RP-HPLC同时测定人体外血浆中7种苯二氮类镇静催眠药的浓度[J].药物分析杂志,2011,31(2):336-339.
[9] Marin SJ, McMillin GA. LC-MS/MS analysis of 13 benzodiazepines and metabolites in urine, serum, plasma, and meconium[J].Methods Mol Biol,2010,603:89-105.
[10]李存,皇甫伟国,杨挺,等.液相色谱串联质谱法测定猪尿中10种镇静剂类药物残留量[J].分析化学研究简报,2010,38(7):1015-1018.
[11]王伟,刘永锁,乔湜,等.UPLC-MS/MS法同时测定人血浆中卡马西平、拉莫三嗪、氯硝西泮、地西泮及其代谢物奥沙西泮浓度[J].中国现代应用药学,2013,30(11):1215-1219.
[12]Sauve EN,Langodegard M,Ekeberg D,et al.Determination of benzodiazepines in ante-mortem and post-mortem whole blood by solid-supported liquid-liquid extraction and UPUPLC-MS/MS[J].J Chromatogr B Analyt Technol Biomed Life Sci,2011,883(884):177-188.
[13]Verplaetse R1,Cuypers E,Tytgat J.The evaluation of the applicability of a high pH mobile phase in ultrahigh performance liquid chromatography tandem mass spectrometry analysis of benzodiazepines and benzodiazepine-like hypnotics in urine and biood [J].J Chromatogr A,2012,12(49):147-154.
猜你喜欢
煤化工(2022年3期)2022-07-08
核化学与放射化学(2022年2期)2022-04-28
色谱(2021年7期)2021-06-07
中国生殖健康(2020年8期)2021-01-18
首都食品与医药(2020年1期)2020-10-21
中国生殖健康(2018年3期)2018-11-06
中国蜂业(2018年4期)2018-05-09
读者·校园版(2017年9期)2017-04-15
山东工业技术(2016年10期)2016-09-06
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
