
Synaptic aging disrupts synaptic morphology and function in cerebellar Purkinje cells
2018-06-21 10:50wenJuanFanMingChaoYanLaiwangYiZhengSunJinBoDengJieXinDeng
中国神经再生研究(英文版) 2018年6期
wen-Juan Fan, Ming-Chao Yan, Lai wang, Yi-Zheng Sun, Jin-Bo Deng, Jie-Xin Deng
Institute of Neurobiology, School of Life Science, Henan University, Kaifeng, Henan Province, China
Introduction
The brain has a very complicated network structure that is composed of neurons and glia, and these neurons are integrated by synaptic connections. A synaptic connection is not only a junction between neurons, but is also the apparatus that transfers an action potential from one neuron to other(Kida and Kato, 2015). Synapses are composed of three elements, a presynaptic membrane, a synaptic cleft and a postsynaptic membrane (Sanders et al., 2015). Dendrites possess many dendritic spines, where the excitatory postsynaptic membrane is located (Amaral and Pozzo-Miller, 2009; Stein et al., 2015). Therefore, dendritic spines are critical for synaptic function and pathogenesis of central nervous system diseases. Synaptic plasticity appears during animal development and continues in aging animals (Lauer et al., 2012; van der Zee, 2015). In our previous study, synaptogenesis was studied in the visual cortex. We showed that nascent synapses could be found as early as embryonic day 15 in the mouse brain. These synapses were structurally immature, with pleiomorphic vesicles in the presumed presynaptic terminal,an unspecialized postsynaptic plasmalemma and a very narrow synaptic cleft. With increasing age, there was gradual thickening of both the pre- and post-synaptic membranes,and the synaptic cleft widened. Up to postnatal day (P14),mature synapse ultrastructure was not seen. The number of dendritic spines and synaptic buttons increased with age and plateaued at P15 (Li et al., 2010). In contrast to synaptic development, synaptic aging is linked with the pathogenesis of some neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease (Arendt et al., 2015; Bate and Williams, 2015). Synaptic damage and synaptic loss are the main manifestations of synaptic aging. Multiple references support the idea that aging disrupts synaptic connections in Alzheimer’s disease (Coleman and Yao, 2003; Baazaoui and Iqbal,2017; Jackson et al., 2017; Baazaoui and Iqbal, 2018). Synapse loss is a key feature of dementia, but it is unclear whether synaptic damage and loss occur in the aging cerebellum.
In this study, we studied synaptic aging of Purkinje cells in the cerebellum. We used immunofluorescent labeling,DiI diolistic assays and transmission electron microscopy, to visualize synaptic buttons, dendritic spines and synaptic ultrastructure of Purkinje cells. Our data will be helpful for understanding the pathogenesis of neurodegenerative diseases.
Materials and Methods
Animals and group assignment
C57BL/6J mice were obtained from the Model Animal Research Center, Nanjing University, China. The experiment was approved by the Animal Ethical Committee of Henan University of China (HUSOM-2016-238; Approval time: 2016-01). Adult male and female C57BL/6 mice were housed in breeding cages with a 12-hour light/dark cycle. P0 was defined as the first 24 hours following birth. Mice were grouped according to the following ages: P0, P3, P7, P14,P30, postnatal month 3 (P3M), P6M, P12M and P18M. A total of 108 male mice were used in this study. Each group contained at least eight mice. Mice at designated ages were intraperitoneally anesthetized with sodium pentobarbital (20 mg/kg), then perfused transcardially with 4% paraformaldehyde in 0.01 M phosphate buffer (pH 7.2). Brains were removed from the skull. Cerebella were carefully separated from brains and fixed in 4% paraformaldehyde for 1–2 days at 4°C.
Immunocytochemistry
Synaptophysin is a synaptic vesicle protein and a marker of presynaptic termini (Feeney et al., 2013). Calbindin is a Purkinje cell-specific marker, (Rajappa et al., 2016). P0 to P18M mice were transcardially perfusion-fixed with 4% paraformaldehyde and postfixed in the same fixative for 1–2 days at 4°C. Sagittal sections (50 µm) were cut with a vibrotome.After rinsing in 0.01 M phosphate buffer, the sections were preincubated in blocking solution (5% normal goat serum and 0.2% Triton-X 100 in 0.01 M phosphate buffer) for 30 minutes at room temperature. The sections were used for synaptophysin/calbindin double immunofluorescence labeling. Primary antibodies for synaptophysin (rabbit anti-synaptophysin polyclonal antibody, 1:300, AB14692; Abcam,Cambridge, MA, USA) and calbindin (mouse anti-calbindin
monoclonal antibody, 1:500, AB82810; Abcam) were added, and the slices were incubated overnight at 4°C. After multiple washes in 0.01 M phosphate buffer, the secondary antibodies, Alexa Fluro 488 donkey ant-rabbit IgG (1:300,A21206; Invitrogen, Carlsbad, CA, USA) and Alexa Fluro 568 donkey anti-mouse IgG (1:600, A10037; Invitrogen)were added and incubated at room temperature for 3 hours.After three washes, sections were mounted in medium (65%glycerol in 0.01 M phosphate buffer + 1:10,000 DAPI for counterstaining). Sections were imaged with an epifluorescence microscope (BX61; Olympus, Tokyo, Japan) under rhodamine, fluorescein isothiocyanate (FITC) or ultraviolet excitation. High-quality sections were photographed with a laser confocal microscope (FV1000; Olympus). The synaptophysin puncta appeared green, and Purkinje cells were stained red with blue nuclei.
1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) diolistic labeling
Diolistic labeling with DiI was performed to visualize the dendritic spines of Purkinje cells. Fixation was the same as for immunocytochemistry, and sagittal cerebellar slices (100µm) were cut using a vibratome. Slices were washed and stored in 0.01 M phosphate buffer for DiI delivery. DiI bullets were prepared according to the method of Li et al. (2010).Briefly, 4 mg of gold particles (1.6 µm diameter) were mixed with 2.5 mg DiI (Sigma) and suspended in 250 µL methylene chloride. The coated particles were immediately transferred to a 1 mm diameter Gene Gun tube (BioRad, Hercules,CA, USA). Gold particles were allowed to adhere to the tube at 4°C for 1–2 days. After withdrawing the solution,the tubing was dried with nitrogen. The tube was cut into small sections. For DiI particle delivery to P0 − P18M mice,slices were transferred to a Petri dish, and the medium was drained. DiI-coated particles were delivered using the Helios Gene Gun system (BioRad) at a pressure of 150 psi. After delivery, slices were incubated in 0.01 M PB (pH 7.2) at 4°C overnight to allow diffusion of the dye along the neuronal processes. After three washes, sections were mounted with 65% glycerol in 0.01 M phosphate buffer.
Transmission electron microscopy
P0 to P18M mice were intraperitoneally anesthetized with sodium pentobarbital (20 mg/kg) and perfused transcardially with 4% paraformaldehyde + 1% glutaraldehyde in phosphate buffer. Cerebella were dissected into tissue blocks of 1.0 mm× 1.0 mm × 1.5 mm. The tissue blocks were post fixed in 4%glutaraldehyde for 4–12 hours at 4°C. After washing several times in phosphate buffer, samples were fixed in 1% OsO4for 1 hour before dehydration in graded ethanol and embedding in Epon 812 resin (Durcupan ACM; Sigma-Aldrich, Gillingham, UK). The Purkinje cell layer was localized in semi-ultrathin sections under light microscopy, and ultrathin 70–80 nm sections were cut (Reichert Ultracut E; Leica, Vienna,Austria) and stained with uranyl acetate, followed by lead citrate. The Purkinje cells and synapses were imaged using an H-7500 electron microscope (Hitachi, Tokyo, Japan).
Photography and confocal imaging
Sections from P0 to P18M mice were photographed with a laser confocal microscope (FV1000; Olympus) using a 100×NA1.4 oil objective and separate laser lines of 356 nm (for nuclei), 488 nm (for synaptic puncta) and 568 nm (for DiI-stained spines and Purkinje cells). Synaptophysin-immunoreactive puncta (green) and DiI-labeled Purkinje cells (red)were visualized using a 4× digital zoom. Both synaptophysin puncta and dendrites were scanned at 0.5 µm intervals along the Z axis. Several images were scanned for dendritic spines and synaptophysin puncta; however, only three neighboring images were overlapped.
Image analysis
The number of synaptophysin puncta and dendritic spine density were measured or calculated using ImageJ analysis software (National Institutes of Health, Bethesda, MD, USA).The following parameters and formulas were used: (1) synapse density = the number of synaptophysin-immunoreactive puncta/area of Purkinje cell layer and molecular layer (puncta/mm2); (2) the density of dendritic spines = the number of dendritic spines/length of dendrites (spines number/µm).
Statistical analysis
Data are expressed as the mean ± SD. The regression equations of age versus synapse density or age versus synaptic buttons were calculated with SPSS 19.0 software (IBM,Armonk, NY, USA). To quantify the aging of synapses and dendritic spines, comparisons among various ages (P3M,P12M and P18M) were made by one-way analysis of variance followed by Student-Newman-Keuls analysis. P3M(young adult mice) was regarded as the control group, but P12M and P18M were regarded as aging groups. A value ofP< 0.05 was considered statistically significant.
Results
Synaptic aging in dendritic spines
DiI diolistic assays and calbindin immunocytochemistry were performed. Purkinje cells could be identified as early as P3. At this age, a row of Purkinje cells appeared between the molecular and granular layers. A bold primary dendrite could be seen extending from the cell body toward the molecular layer, and two or three short dendrites branched from the primary dendrite. An axon from each Purkinje cell penetrated through the granular layer and entered into the white matter of the cerebellum (Figure 1D). In this way,the molecular layer was filled with a few short Purkinje cells dendrites. With increasing age, the Purkinje cells grew,and dendrites branched repeatedly. Finally, the Purkinje cells appeared fan-shaped with numerous dendrites in the molecular layer (Figures 1B, 2A, 2B). At P30, the Purkinje cells dendrites reached the pia (Figure 1B). The DiI diolistic assay showed dendritic spines on dendrites. At P3, dendritic spines started to sprout, and many of them looked like filopodia (Figure 1D). With dendrite growth into the molecular layer, the number of dendritic spines rapidly increased (Figure 1A, E). At P14, the spine density increased further, and the spines became short and neatly arranged (Figure 1F). At P30, the spine density peaked and remained at this level until P12M (Figures 1B, 1G, 3A). At one year of age, dendritic spines started to show alterations to their shape and quantity. The dendritic spine density gradually decreased (Figures 1C, 1I, 3A). After curve fitting between spine density (Y)and age (X), regression was calculated byY= 0.025X3−0.416X2+ 2.172X− 1.586 (R² = 0.989;P< 0.05). Compared with control (P3M), dendritic spines were gradually lost at P12M and P18M (P< 0.05).
Quantity and ultrastructure of aging synapses
Synaptophysin is specifically expressed in synaptic vesicles of presynaptic terminals. Therefore, synaptophysin can be used as a marker for presynaptic terminals, and the number of synaptophysin-immunoreactive puncta represents the number of synapses. This study used calbindin and synaptophysin to co-label Purkinje cells and presynaptic terminals(Figure 2A−D). Synaptophysin-immunoreactive puncta were usually located on spines, dendrites and cell bodies of Purkinje cells, suggesting that they represented synaptic connections on Purkinje cells. At P3, only a few synaptophysin-immunoreactive puncta were seen around the dendrites and cell bodies of Purkinje cells. Only a few synaptophysin-immunoreactive puncta were seen in the molecular layer, because there were no Purkinje cell dendrites in this layer (Figure 2A). At P7, the number of synaptophysin-immunoreactive puncta increased, and many synaptophysin-immunoreactive puncta were located in the lower molecular layer because the Purkinje cells’ dendrites extended into the molecular layer (Figure 2B). At P30, the number of synaptophysin-immunoreactive puncta peaked they were distributed throughout the Purkinje cell and molecular layers (Figures 2C, 3B). The number of synaptophysin-immunoreactive puncta remained at this plateau level until P12M.The number of synaptophysin-immunoreactive puncta then decreased and reached a minimum after P18M (P< 0.05;Figures 2D, 3B). After fitting the curve between the synaptic buttons (Y) and age (X), regression was calculated byY=0.013X3− 0.244X2+ 1.384X− 0.973 (R² = 0.972,P< 0.05)(Figure 3B). Compared with control (P3M), synaptic button numbers at P12M and P18M decreased gradually (P< 0.05).These results confirm that synapse loss occurs during synaptic aging.
Synaptic ultrastructure was observed by transmission electron microscopy. As early as P7, the ultrastructure of synapses could be observed with a presynaptic membrane,a synaptic cleft (25–30 nm) and a postsynaptic membrane(Figure 4A). At P30, the synaptic structure was mature, with presynaptic terminals containing mitochondria and a large number of synaptic vesicles (33–36 nm) (Figure 4B). Both presynaptic and postsynaptic membranes had thickened to form the specialized zones (Figure 4C). After P12M, synapses started to age. The numbers of synaptic vesicles and mitochondria in presynaptic terminals were reduced, and both presynaptic and postsynaptic membranes became thin(Figure 4C, D).
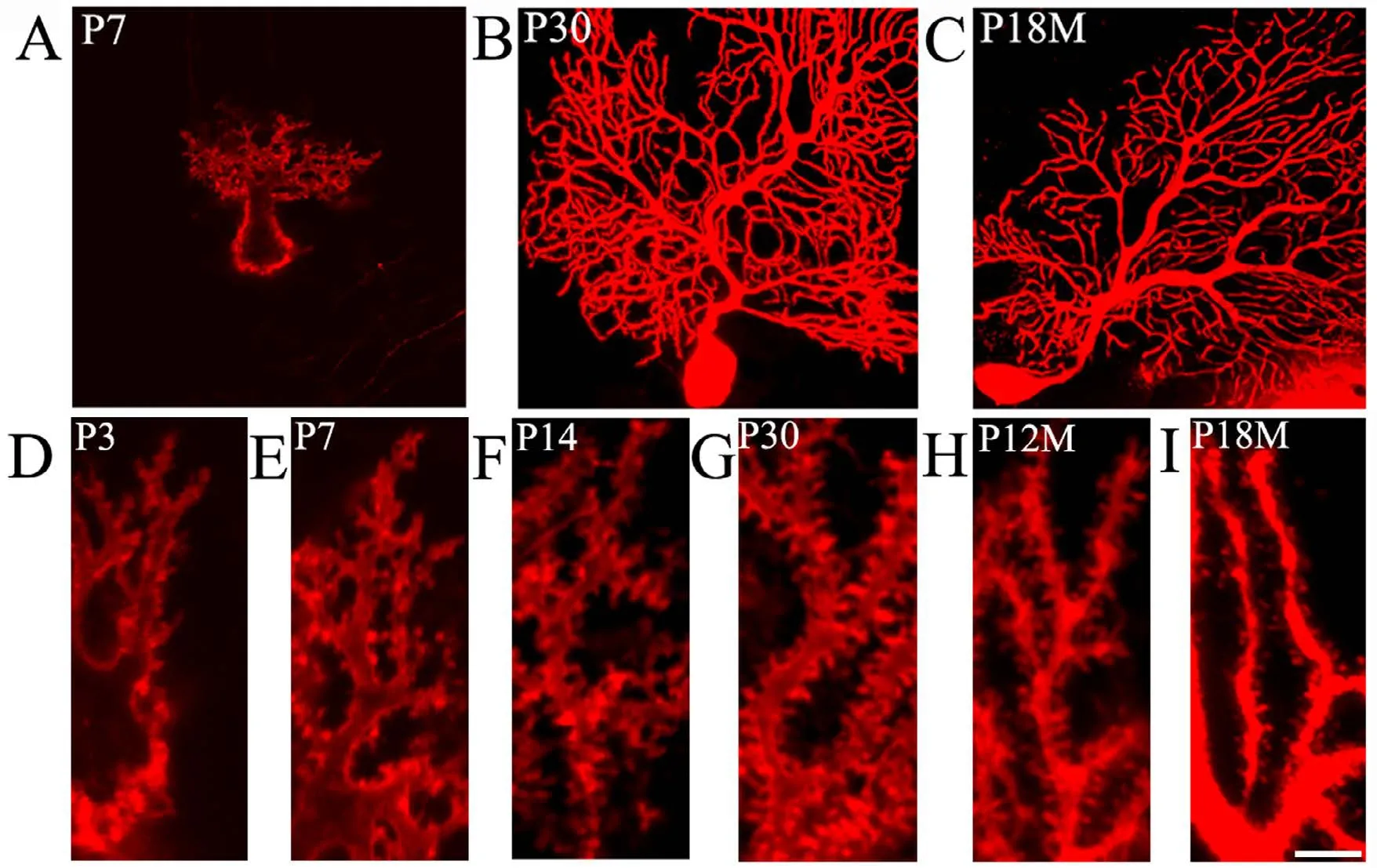
Figure 1 Aging of dendritic spines in Purkinje cells (DiI diolistic assay).
Discussion
Dendritic spines and synaptic buttons are lost during synaptic aging
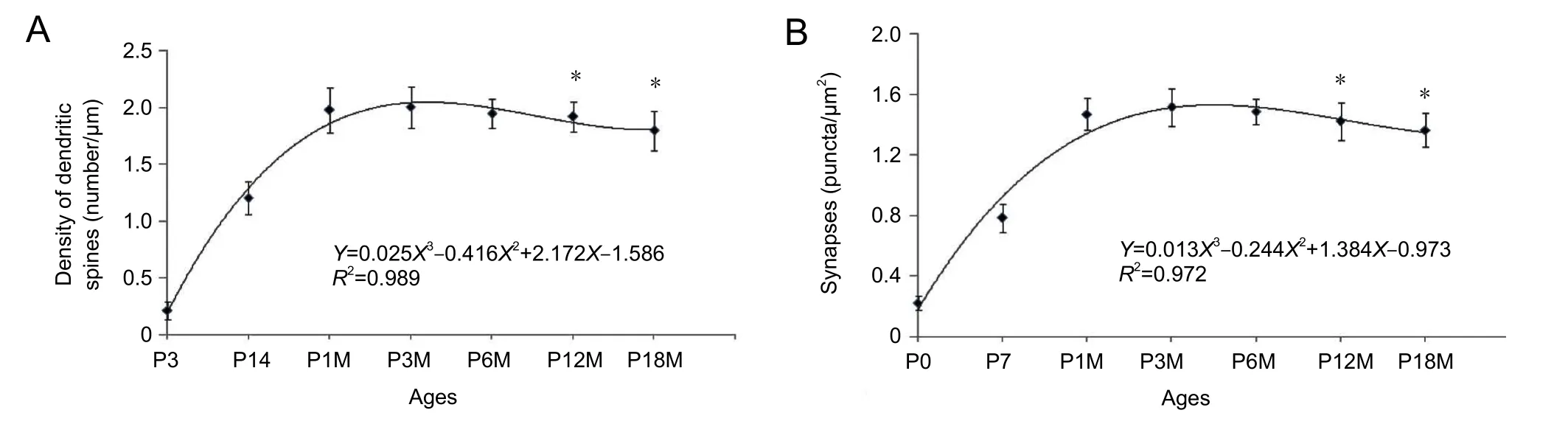
Figure 3 Changes in the density of dendritic spines and synaptic buttons in Purkinje cells during synaptic aging.
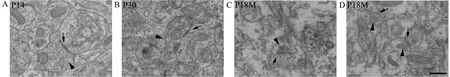
Figure 4 Synaptic ultrastructure during synaptic development and aging (transmission electron microscope).
The synapse is the key structure in neural networks that transduces action potentials between neurons. Synapses are crucial to learning and memory in the central nervous system (Whalley, 2014; Wu et al., 2017). Therefore, studies of synaptogenesis and synaptic aging are important for understanding neural development and neural pathogenesis.Our previous study found that synaptogenesis and synaptic maturation were harmonized with brain development and maturation (Li et al., 2010). Many neurodegenerative diseases are associated with disorders of synaptic function, such as Alzheimer disease (Toth et al., 2017; Rajendran and Paolicelli, 2018). Pathological Aβ strongly inhibits the synaptic vesicle fusion machinery (An et al., 2013; Yuyama and Igarashi, 2017). In Parkinson disease, pathological mutations of Parkinson disease factors result in abnormal degradation of synaptic proteins (Collins et al., 2015; Morales et al., 2015).Therefore, the study of synaptic aging is significant for basic theoretical understanding and clinic practice. This study sought to understand the relationship between synaptic aging and quantitative alterations of synapses. Synaptophysin is a calcium binding acidic glycoprotein located around synaptic vesicles, and it plays an important role in neurotransmitter release (Mallozzi et al., 2013; Gordon et al.,2016). Dendritic spines can also be used to reflect the state of synapses, because the postsynaptic element of excitatory synapse is often located on spines (Sasaki et al., 2014; Lei et al., 2017). In the present study, synaptophysin immunocytochemistry and Dil diolistic assays were used to visualize synaptic buttons and dendritic spines, as in our previous experiment (Li et al., 2010), and the number of synaptic buttons and dendritic spines on Purkinje cells were investigated during synaptic aging.
Purkinje cell dendrites can receive information from parallel fibers in the molecular layer; therefore, numerous synapses exist on Purkinje cell dendrites (Lippiello et al.,2015). Our results showed that in adult mice from P30 to P12M, the synapse density remained at a plateau level. The synapses started to show aging after P12M, when the action of synaptophysin-immunoreactive synaptic buttons begins to slow (Kommaddi et al., 2017). These phenotypic characteristics may result from Purkinje cell damage (Cheng et al., 2011). Cell apoptosis leads to morphological changes of Purkinje cells with reduced numbers of dendrites and spines and even synaptic disassembly (Zhang et al., 2006; Li et al.,2013). Cell apoptosis also reduces neurotrophic influence on Purkinje cells, and causes further dendritic reduction (Li et al., 2013). The cytoskeleton is involved in the plasticity of dendritic spines (Badura et al., 2013; Bellot et al., 2014; Min et al., 2017). With neural aging, the cytoskeleton in dendritic spines starts to disassemble (Young et al., 2014), resulting in the retraction of dendritic spines (Shimada et al., 2006).Therefore, during synaptic aging, dendritic spines and synapses are gradually lost.
Ultrastructural changes during synaptic aging
Synaptic aging refers to morphological and functional degeneration, and even disassembly of the synapse (Weber et al., 2015; Smidak et al., 2017). The study of synaptic aging is important for understanding the pathogenesis of neural degenerative diseases (Foster et al., 2017). The ultrastructure of a synapse consists of a presynaptic element, a synaptic cleft and a postsynaptic element (Tremblay et al., 2013).Ultrastructural alterations strongly affect synaptic function;therefore, synaptic aging will affect the transduction of action potentials, and learning and memory (Lee and Littleton, 2015; Shetty et al., 2017; Todorova and Blokland, 2017).In this study, using transmission electron microscopy, the ultrastructure of aging synapses was systematically investigated. After P12M, the numbers of synaptic vesicles and mitochondria in presynaptic terminals were reduced, and the specialized zones of pre- and post-synaptic membranes became thin. Synaptic vesicles store neurotransmitters (Lee and Littleton, 2015), and mitochondria provide energy for neurotransmitter release (Hrynevich et al., 2015; Lee and Littleton, 2015). In aged synapses, because of the reduced number of synaptic vesicles and mitochondria, neurotransmitter release may not occur normally, resulting in impaired neural signal transduction (Luo et al., 2014). Furthermore,the postsynaptic membrane contains many neurotransmitter receptors. The thin postsynaptic membrane in an aged synapse is likely to cause these receptors to malfunction (Nair et al., 2013). Aged synapses are often found in neurodegenerated brains, such as in Alzheimer disease and Parkinson disease (Whit field et al., 2015). Norepinephrine is involved in synaptic aging, especially in the cerebellum. Norepinephrine can regulate Purkinje cell activity with the cooperation of γ-aminobutyric acid (Bickford, 1993). In aging Purkinje cells, cell activity decreases because of insufficient norepinephrine in the synapse (Bickford-Wimer et al., 1988; Louis et al., 2009). Subsequently, levels of neurotransmitter hydrolases decrease, which weakens the activity of glutamate dehydrogenase, causing glutamate accumulation. The accumulated glutamate is toxic to neurons and can cause further neuronal apoptosis (Rossi et al., 2006; Fu et al., 2011; Morland et al., 2016).
以上4种土地利用分级是在理想状态下划分的,但在实际状态中,这4种土地利用分级都分布在同一地区,有不同的面积比例并根据其权重对这一地区的土地利用变化产生不同的影响。根据刘纪远提出的土地利用程度综合分析方法,形成了一个在不同土地利用分级之间连续分布的综合指数,可反映某一地区的土地利用程度,即土地利用程度综合指数。以土地利用程度综合指数为基础反映土地利用程度差异的模型即土地利用程度变化模型,其指标及模型含义如下:
In summary, dendritic spines and synaptic buttons are lost during synaptic aging. Simultaneously, aged synapses also exhibit ultrastructural alterations, such as a reduction in the number of synaptic vesicles and mitochondria in presynaptic terminals and thin pre- and post-synaptic membranes.Aging synapses can impede neurotransmitter release, resulting in disordered synaptic morphology and function.
Author contributions:LW and JBD designed the study. WJF and MCY performed most experiments and analyzed data. WJF wrote and edited the paper. YZS provided technical support. JXD participated in statistical processing. All authors approved the final version of the paper.
Conflicts of interest:The authors declare no competing financial interests.Financial support:This research was supported by the Science and Technology Projects of Henan Province of China, No. 172102310001;the Biology Advantage Discipline Fund of Henan Province of China. The funders did not participant in the study design, in the collection, analysis and interpretation of data, in the writing of the paper, and in the decision to submit the paper for publication.
Institutional review board statement:The animal usage in the experiment was approved by Animal Ethical Committee of Henan University of China (HUSOM-2016-238; Approval time: 2016-01). The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1985).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Dirk Montag, Head Neurogenetics Special Laboratory, Leibniz Institute for Neurobiology, Neurogenetics Special Laboratory, Germany; Ivan Fernandez-Vega, Hospital Universitario Central de Asturias, Spain.
Additional file:Open peer review reports 1 and 2.
Amaral MD, Pozzo-Miller L (2009) The dynamics of excitatory synapse formation on dendritic spines. Cellscience 5:19-25.
An K, Klyubin I, Kim Y, Jung JH, Mably AJ, O’Dowd ST, Lynch T,Kanmert D, Lemere CA, Finan GM, Park JW, Kim TW, Walsh DM,Rowan MJ, Kim JH (2013) Exosomes neutralize synaptic-plasticity-disrupting activity of Abeta assemblies in vivo. Mol Brain 6:47.
Arendt KL, Zhang Z, Ganesan S, Hintze M, Shin MM, Tang Y, Cho A,Graef IA, Chen L (2015) Calcineurin mediates homeostatic synaptic plasticity by regulating retinoic acid synthesis. Proc Natl Acad Sci U S A 112:E5744-E5752.
Baazaoui N, Iqbal K (2017) Prevention of dendritic and synaptic de ficits and cognitive impairment with a neurotrophic compound. Alzheimers Res Ther 9:45.
Baazaoui N, Iqbal K (2018) A novel therapeutic approach to treat Alzheimer’s disease by neurotrophic support during the period of synaptic compensation. J Alzheimers Dis 62:1211-1218.
Badura A, Schonewille M, Voges K, Galliano E, Renier N, Gao Z, Witter L, Hoebeek FE, Chedotal A, De Zeeuw CI (2013) Climbing fiber input shapes reciprocity of Purkinje cell firing. Neuron 78:700-713.
Bate C, Williams A (2015) alpha-Synuclein-induced synapse damage in cultured neurons is mediated by cholesterol-sensitive activation of cytoplasmic phospholipase A2. Biomolecules 5:178-193.
Bellot A, Guivernau B, Tajes M, Bosch-Morato M, Valls-Comamala V,Munoz FJ (2014) The structure and function of actin cytoskeleton in mature glutamatergic dendritic spines. Brain Res 1573:1-16.
Bickford P (1993) Motor learning deficits in aged rats are correlated with loss of cerebellar noradrenergic function. Brain Res 620:133-138.
Bickford-Wimer PC, Granholm AC, Gerhardt GA (1988) Cerebellar noradrenergic systems in aging: studies in situ and in in oculo grafts.Neurobiol Aging 9:591-599.
Cheng XS, Li MS, Du J, Jiang QY, Wang L, Yan SY, Yu DM, Deng JB(2011) Neuronal apoptosis in the developing cerebellum. Anat Histol Embryol 40:21-27.
Coleman PD, Yao PJ (2003) Synaptic slaughter in Alzheimer’s disease.Neurobiol Aging 24:1023-1027.
Collins JM, King AE, Woodhouse A, Kirkcaldie MT, Vickers JC (2015)The effect of focal brain injury on beta-amyloid plaque deposition,inflammation and synapses in the APP/PS1 mouse model of Alzheimer’s disease. Exp Neurol 267:219-229.
Feeney EJ, Stephenson D, Kleiman R, Bove S, Cron C, Moody L, Robinson M, Ramirez JJ (2013) Immunohistochemical characterization of axonal sprouting in mice. Restor Neurol Neurosci 31:517-531.
Foster TC, Kyritsopoulos C, Kumar A (2017) Central role for NMDA receptors in redox mediated impairment of synaptic function during aging and Alzheimer’s disease. Behav Brain Res 322:223-232.
Fu L, Tang R, Bao N, Wang J, Ma H (2011) Ketamine and propofol in combination induce neuroapoptosis and down-regulate the expression of N-methyl-D-aspartate glutamate receptor NR2B subunit in rat forebrain culture. Pharmazie 66:771-776.
Gordon SL, Harper CB, Smillie KJ, Cousin MA (2016) A fine balance of synaptophysin levels underlies efficient retrieval of synaptobrevin II to synaptic vesicles. PLoS One 11:e0149457.
Hrynevich SV, Pekun TG, Waseem TV, Fedorovich SV (2015) Influence of glucose deprivation on membrane potentials of plasma membranes, mitochondria and synaptic vesicles in rat brain synaptosomes. Neurochem Res 40:1188-1196.
Jackson JS, Witton J, Johnson JD, Ahmed Z, Ward M, Randall AD,Hutton ML, Isaac JT, O’Neill MJ, Ashby MC (2017) Altered synapse stability in the early stages of tauopathy. Cell Rep 18:3063-3068.
Kida S, Kato T (2015) Microendophenotypes of psychiatric disorders:phenotypes of psychiatric disorders at the level of molecular dynamics, synapses, neurons, and neural circuits. Curr Mol Med 15:111-118.
Kommaddi RP, Das D, Karunakaran S, Nanguneri S, Bapat D, Ray A,Shaw E, Bennett DA, Nair D, Ravindranath V (2017) Abeta mediates F-actin disassembly in dendritic spines leading to cognitive deficits in Alzheimer’s disease. J Neurosci 38:1085-1099.
Lauer AM, Fuchs PA, Ryugo DK, Francis HW (2012) Efferent synapses return to inner hair cells in the aging cochlea. Neurobiol Aging 33:2892-2902.
Lee J, Littleton JT (2015) Transmembrane tethering of synaptotagmin to synaptic vesicles controls multiple modes of neurotransmitter release. Proc Natl Acad Sci U S A 112:3793-3798.
Lei W, Myers KR, Rui Y, Hladyshau S, Tsygankov D, Zheng JQ (2017)Phosphoinositide-dependent enrichment of actin monomers in dendritic spines regulates synapse development and plasticity. J Cell Biol 216:2551-2564.
Li J, Yu L, Gu X, Ma Y, Pasqualini R, Arap W, Snyder EY, Sidman RL(2013) Tissue plasminogen activator regulates Purkinje neuron development and survival. Proc Natl Acad Sci U S A 110:E2410-E2419.Li M, Cui Z, Niu Y, Liu B, Fan W, Yu D, Deng J (2010) Synaptogenesis in the developing mouse visual cortex. Brain Resea Bull 81:107-113.
Lippiello P, Hoxha E, Volpicelli F, Lo Duca G, Tempia F, Miniaci MC(2015) Noradrenergic modulation of the parallel fiber-Purkinje cell synapse in mouse cerebellum. Neuropharmacology 89:33-42.
Louis ED, Faust PL, Vonsattel JP, Erickson-Davis C (2009) Purkinje cell axonal torpedoes are unrelated to advanced aging and likely reflect cerebellar injury. Acta Neuropathol 117:719-721.
Luo F, Guo NN, Li SH, Tang H, Liu Y, Zhang Y (2014) Reduction of glutamate release probability and the number of releasable vesicles are required for suppression of glutamatergic transmission by beta1-adrenoceptors in the medial prefrontal cortex. Neuropharmacology 83:89-98.
Mallozzi C, D’Amore C, Camerini S, Macchia G, Crescenzi M, Petrucci TC, Di Stasi AM (2013) Phosphorylation and nitration of tyrosine residues affect functional properties of Synaptophysin and Dynamin I, two proteins involved in exo-endocytosis of synaptic vesicles. Biochim Biophys Acta 1833:110-121.
Min H, Dong J, Wang Y, Wang Y, Yu Y, Shan Z, Xi Q, Teng W, Chen J (2017) Marginal iodine deficiency affects dendritic spine development by disturbing the function of rac1 signaling pathway on cytoskeleton. Mol Neurobiol 54:437-449.
Morales I, Sanchez A, Rodriguez-Sabate C, Rodriguez M (2015) The degeneration of dopaminergic synapses in Parkinson’s disease: a selective animal model. Behav Brain Res 289:19-28.
Morland C, Pettersen MN, Hassel B (2016) Hyperosmolar sodium chloride is toxic to cultured neurons and causes reduction of glucose metabolism and ATP levels, an increase in glutamate uptake, and a reduction in cytosolic calcium. Neurotoxicology 54:34-43.
Nair R, Lauks J, Jung S, Cooke NE, de Wit H, Brose N, Kilimann MW,Verhage M, Rhee J (2013) Neurobeachin regulates neurotransmitter receptor trafficking to synapses. J Cell Biol 200:61-80.
Rajappa R, Gauthier-Kemper A, Boning D, Huve J, Klingauf J (2016)Synaptophysin 1 clears synaptobrevin 2 from the presynaptic active zone to prevent short-term depression. Cell Rep 14:1369-1381.
Rajendran L, Paolicelli RC (2018) Microglia-mediated synapse loss in Alzheimer’s disease. J Neurosci 38:2911-2919.
Rossi S, Prosperetti C, Picconi B, De Chiara V, Mataluni G, Bernardi G,Calabresi P, Centonze D (2006) Deficits of glutamate transmission in the striatum of toxic and genetic models of Huntington’s disease.Neurosci Lett 410:6-10.
Sanders J, Singh A, Sterne G, Ye B, Zhou J (2015) Learning-guided automatic three dimensional synapse quantification for drosophila neurons. BMC Bioinformatics 16:177.
Sasaki T, Oga T, Nakagaki K, Sakai K, Sumida K, Hoshino K, Miyawa
ki I, Saito K, Suto F, Ichinohe N (2014) Developmental expression profiles of axon guidance signaling and the immune system in the marmoset cortex: potential molecular mechanisms of pruning of dendritic spines during primate synapse formation in late infancy and prepuberty (I). Biochem Biophysical Res Commun 444:302-306.Shetty MS, Sharma M, Sajikumar S (2017) Chelation of hippocampal zinc enhances long-term potentiation and synaptic tagging/capture in CA1 pyramidal neurons of aged rats: implications to aging and memory. Aging Cell 16:136-148.
Shimada A, Tsuzuki M, Keino H, Satoh M, Chiba Y, Saitoh Y, Hosokawa M (2006) Apical vulnerability to dendritic retraction in prefrontal neurones of ageing SAMP10 mouse: a model of cerebral degeneration. Neuropathol Appl Neurobiol 32:1-14.
Smidak R, Sialana FJ, Kristofova M, Stojanovic T, Rajcic D, Malikovic J, Feyissa DD, Korz V, Hoeger H, Wackerlig J, Mechtcheriakova D,Lubec G (2017) Reduced levels of the synaptic functional regulator FMRP in dentate gyrus of the aging sprague-dawley rat. Front Aging Neurosci 9:384.
Stein IS, Gray JA, Zito K (2015) Non-Ionotropic NMDA receptor signaling drives activity-induced dendritic spine shrinkage. J Neuroscience 35:12303-12308.
Todorova V, Blokland A (2017) Mitochondria and synaptic plasticity in the mature and aging nervous system. Curr Neuropharmacol 15:166-173.
Toth P, Tarantini S, Csiszar A, Ungvari Z (2017) Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol 312:H1-H20.
Tremblay ME, Marker DF, Puccini JM, Muly EC, Lu SM, Gelbard HA(2013) Ultrastructure of microglia-synapse interactions in the HIV-1 Tat-injected murine central nervous system. Commun Integr Biol 6:e27670.
van der Zee EA (2015) Synapses, spines and kinases in mammalian learning and memory, and the impact of aging. Neurosci Biobehav Rev 50:77-85.
Weber M, Wu T, Hanson JE, Alam NM, Solanoy H, Ngu H, Lauffer BE, Lin HH, Dominguez SL, Reeder J, Tom J, Steiner P, Foreman O, Prusky GT, Scearce-Levie K (2015) Cognitive deficits, changes in synaptic function, and brain pathology in a mouse model of normal aging (1,2,3). eNeuro doi: 10.1523/ENEURO.0047-15.2015.
Whalley K (2014) Learning and memory: synapse remodelling extinguishes fear. Nat Rev Neurosci 15:3.
Whitfield DR, Vallortigara J, Alghamdi A, Hortobagyi T, Ballard C,Thomas AJ, O’Brien JT, Aarsland D, Francis PT (2015) Depression and synaptic zinc regulation in Alzheimer disease, dementia with lewy bodies, and Parkinson disease dementia. Am J Geriatr Psychiatry 23:141-148.
Wu C, Kim TW, Choi HY, Strukov DB, Yang JJ (2017) Flexible three-dimensional artificial synapse networks with correlated learning and trainable memory capability. Nat Commun 8:752.
Young ME, Ohm DT, Dumitriu D, Rapp PR, Morrison JH (2014)Differential effects of aging on dendritic spines in visual cortex and prefrontal cortex of the rhesus monkey. Neuroscience 274:33-43.
Yuyama K, Igarashi Y (2017) Exosomes as Carriers of Alzheimer’s Amyloid-ss. Front Neurosci 11:229.
Zhang C, Hua T, Zhu Z, Luo X (2006) Age-related changes of structures in cerebellar cortex of cat. J Biosci 31:55-60.
猜你喜欢
成都信息工程大学学报(2022年2期)2022-06-14
资源信息与工程(2021年5期)2022-01-15
趣味(语文)(2021年10期)2021-12-28
农业资源与环境学报(2021年4期)2021-07-30
家庭影院技术(2021年5期)2021-07-21
建材发展导向(2021年10期)2021-07-16
趣味(作文与阅读)(2021年10期)2021-03-08
意林(2021年2期)2021-02-08
中学生英语·阅读与写作(2014年11期)2015-03-11
中学生英语·阅读与写作(2014年3期)2014-07-29

- 中国神经再生研究(英文版)的其它文章
- Role of brain-derived neurotrophic factor during the regenerative response after traumatic brain injury in adult zebrafish
- Natural polyphenols effects on protein aggregates in Alzheimer’s and Parkinson’s prion-like diseases
- How random is the random forest ? Random forest algorithm on the service of structural imaging biomarkers for Alzheimer’s disease: from Alzheimer’s disease neuroimaging initiative (ADNI) database
- Protective effects of gonadal hormones on spinal motoneurons following spinal cord injury
- INVITED REVIEW
- Role of presynaptic calcium stores for neural network dysfunction in Alzheimer’s disease