
食品中99种兽药的一步式提取净化体系
2018-06-20 06:42:52陈树兵李露青徐旭文陈先锋钟莺莺张雅珩王传现
色谱 2018年6期
李 双, 陈树兵, 李露青, 徐旭文,陈先锋, 钟莺莺, 张雅珩, 王传现
(1. 宁波出入境检验检疫局检验检疫技术中心, 浙江 宁波 315211; 2. 宁波大学应用海洋生物技术教育部重点实验室, 浙江 宁波 315211; 3. 甘肃出入境检验检疫局综合技术中心, 甘肃 兰州 730010; 4. 上海出入境检验检疫局, 上海 200135)
禁限用兽药超量和超种类的滥用是目前食品安全所面对的重大问题之一。近年来,由于养殖条件差异和利益驱使,兽药超标现象时有发生,激素类残留非常普遍,阳性样品检出率逐年攀升[1,2]。原料鸡养殖过程中所用抗生素种类繁多,甚至抗生素停药期不停用,使用国家禁用的地塞米松类药物[3]。目前,我国兽药残留检测的标准多达数十个,多是按照某一类兽药建立的,且适应食品基质单一[4-7]。因此,建立食品中多兽药的高通量筛查方法,对提高监管工作效率、保障食品安全具有重要的意义。
兽药检测主要涉及两方面技术:前处理技术和仪器分析。常见的前处理技术包括固相萃取、固相微萃取、基质分散固相萃取、免疫亲和色谱技术、分子印迹技术、超临界流体萃取、加速溶剂萃取等[8,9]。这些前处理技术各有特点,但都存在选择性限制的弊端,样品处理通量不高。例如,于洁等[10]对16种基质中120种兽药同时筛查,前处理采用多次分步提取净化步骤;张晓光等[11]检测保健食品中的激素类物质,用乙腈作为提取液,没有兼容强极性物质的提取;同样地,新兴的QuEChERS净化方法也存在上述问题,不能保证强极性化合物的回收率[12]。如何把极性物质从水溶液中有效提取出来,这始终是一个艰巨的挑战[13]。
随着质谱的发展和推广,一次进样完成多类残留分析已成为可能。兽药残留正逐渐由液相色谱-三重四极杆质谱(LC-MS/MS)目标型检测向高分辨质谱(HRMS)精确质量非目标型全扫描检测转变[14-19]。传统的LC-MS/MS在定量分析上具有优势,但分析化合物数量有限,只能针对方法中涵盖的物质,并需要逐个进行参数优化,耗时且对基质干扰敏感,相应的前处理也就更为复杂;而且分辨率低,不能有效区分相对分子质量相近的化合物,会造成假阳性结果等。全扫描高分辨质谱仪则不需要待测化合物的特征离子碎片,直接采集高准确度质量数(分辨率在70 000以上),对待测样品进行全扫描,需要增加目标化合物时,不必再次进样,重新分析已有的全扫描数据即可。在高分辨率下采集的数据降低了近似质量数的干扰,更好地避免了假阳性的发生。通过多级扫描,建立二级谱图数据库,特别适合高通量的检测筛查。
本文利用载体辅助液液萃取技术,通过一次前处理,涵盖了常见的理化性质差异很大的8大类共计99种兽药残留的提取、净化工作,同时结合四极杆静电场轨道阱高分辨质谱,实现了畜类、禽类、液态乳、奶粉、鱼类等动物源性食品中常见兽药残留的一步式多残留筛查,为提高政府监管部门的监管工作效率,保证动物源性食品安全提供了基础。
1 实验部分
1.1 仪器、试剂与材料
Q-Exactive四极杆静电场轨道阱高分辨质谱仪(赛默飞世尔科技公司),配有H-ESI II源。液相色谱系统为UltiMate 3000高压液相色谱,配有自动进样器。色谱柱为Thermo Hypersil Gold C18柱(100 mm×2.1 mm, 1.9 μm)。Milli-Q高纯水发生器(美国Millipore公司)。冷冻干燥机(美国LABCONCO公司);旋转蒸发仪(德国IKA公司)。
兽药标准品购自美国Sigma-Aldrich公司和德国Dr. Ehrenstorfer公司,纯度≥95%。基质样品来自国家植物源性残留监控抽样和进出口送检企业。硫酸铵、二甲基亚砜、乙二胺四乙酸二钠(EDTA)、氨水(均为分析纯)购自南京化学试剂一厂;色谱纯甲酸购自美国Sigma-Aldrich公司。其他试剂均为色谱纯,购自德国Merck公司。实验用水为Milli-Q超纯水(18.2 ΩM5cm)。
1.2 标准溶液的配制
准确称取10 mg标准品,用乙腈稀释成1 000 mg/L的标准储备液;将标准物质分为3组,配制混标,第一组为28种激素类化合物,第二组为39种磺胺喹诺酮类化合物,第三组为32种其他兽药;分别准确吸取100 μL标准储备液,用乙腈溶液定容至10 mL,配制10 mg/L混合标准溶液,于-20 ℃避光保存。
1.3 一步式提取净化体系操作
提取液的配制:称取4.3 g的草酸和3.7 g EDTA,用500 mL水溶解,氨水调pH至3.0; 100 mmol/L草酸缓冲液的配制:称取0.9 g草酸,用90 mL水溶解,氨水调pH至5.0,加水至100 mL。
称取5.00 g样品,加入5 mL提取液和15 mL乙腈,振荡5 min, 4 500 r/min下离心5 min;取出上清液,加入1 g硫酸铵,振荡5 min, 4 500 r/min下离心5 min,得到乙腈层(上)和水层(下);取水层上样,平衡15 min,乙腈作为洗脱液进行洗脱,再依次用10 mL乙腈清洗离心管2次,并依次上柱洗脱,下接50 mL离心管,并加入0.5 mL二甲基亚砜,乙腈定容至25 mL;取出12.5 mL,加入200 μL草酸缓冲液,氮吹后用纯水定容至1.0 mL,过0.22 μm滤膜,高浓度的加标水平稀释到线性范围内,待分析。
1.4 色谱及质谱条件
液相色谱条件:色谱柱Hypersil Gold C18 (100 mm×2.1 mm 1.9 μm),部分激素成分(ESI-)的流动相A:含有0.1%(v/v)氨水的乙腈溶液;流动相B:含有0.1% (v/v)氨水的水溶液;混合标准溶液中其余成分(ESI+)的流动相A:含有0.1%(v/v)甲酸和5 mmol/L乙酸铵的乙腈溶液,流动相B:含有0.1%(v/v)甲酸和5 mmol/L乙酸铵的水溶液。流速:0.3 mL/min,进样量:10 μL,梯度洗脱程序见表1。
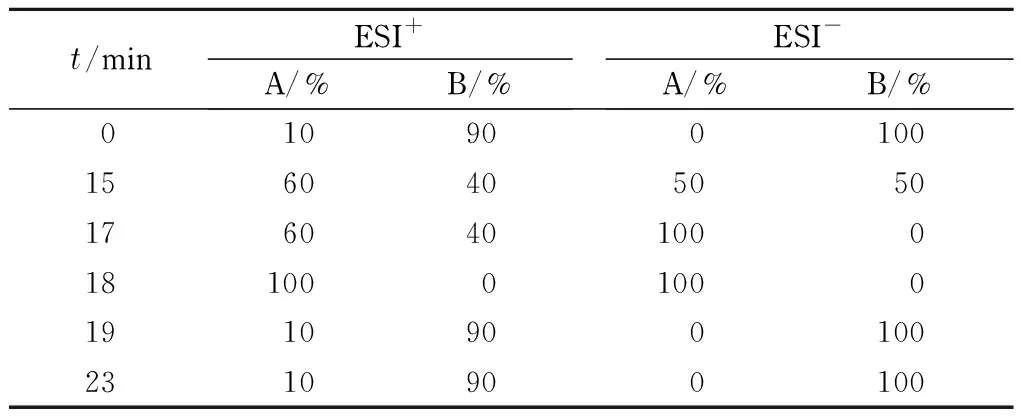
表 1 ESI+和ESI-模式下的HPLC洗脱程序
质谱条件:质谱在正/负离子转换模式下进行全扫描测定,质量范围:m/z100~1 000,分辨率70 000,自动增益控制(AGC)目标值5×105;部分激素成分采用负离子模式2 700 V,其余成分则采用正离子模式3 800 V,离子传输管温度为300 ℃,鞘气压(N2)35 arb,辅助气压(N2)10 arb,气化室温度350 ℃;在样品运行前对仪器分别进行正、负离子校正;二级采用自动触发模式,分辨率35 000, AGC目标值2×105,碰撞能量范围25%~40%,保留时间采集范围:根据一级色谱图中各个目标物质的保留时间(RT)±1.00 min。
1.5 兽药的定性、定量分析
定性分析:精确质量误差低于5×10-6,同时比对保留时间、同位素分布、主要二级碎片和二级质谱图相似度,综合判断后得到准确定性结果,避免假阳性结果出现。部分标准物质图谱见图1。
定量分析:用3组10 mg/L混合标准溶液配制系列浓度的标准溶液:0、1.0、2.0、5.0、10、20、50、100 ng/mL,以质量浓度为横坐标,标准品峰面积为纵坐标,做基质标准曲线,作为待测物定量的依据。部分标准物质在牛奶、猪肉、虾基质中的线性方程和相关系数见附表1(http://www.chrom-china.com/UserFiles/File/SP1802023SI.pdf)。
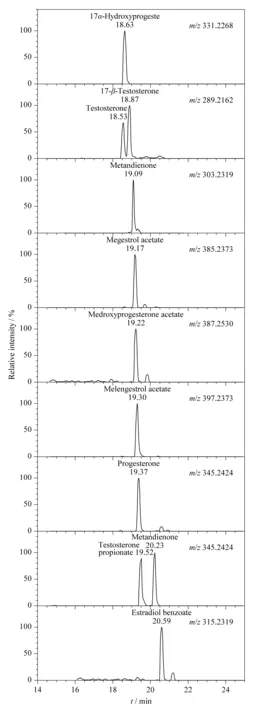
图 1 部分兽药标准物质的色谱图Fig. 1 Chromatograms of some veterinary drugs
2 结果与讨论
2.1 提取液的选择
液体样品或者固体水样首先经过强极性水性提取液进行提取,可得到水溶性等强极性的兽药,同时避免脂肪类等低极性物质的干扰。提取液中含有的EDTA也可以很好地络合奶制品中的大量金属离子;然后加入3倍体积的乙腈(15 mL),沉淀样品水提液中的蛋白质组分。乙腈溶解性好、渗透强,已经成为食品基质中常用的提取溶剂,它同时可以防止极性相对偏弱的兽药随蛋白质沉淀而损失。
2.2 提取剂的添加量
提取液的添加量要充分考虑柱子的填料含量,本文选择的硅藻土柱型号为Chromabond XTR (1 000 mg),分别比较了上样量为5、8、10、12、15 mL时的柱负载,在上样量达到12 mL和15 mL的时候,柱已达到饱和并渗出,水溶液的渗出会导致最后的氮吹浓缩过程大大延长。另外,当上样量为5 mL和8 mL的时候,柱填料未充分浸入水溶液,大大降低了柱子的利用率,同时对于兽药本就很低的检出限量来说,也增加了仪器检出的难度。因此,本研究最终将上样体积确定为10 mL。
2.3 辅助提取剂乙腈的添加量
在分级提取过程中,乙腈作为水的辅助提取剂,其加入量对不同极性兽药的提取率有很大的影响。向液体基质牛奶和固体基质猪肉样品中添加99种禁限用兽药,通过加入不同体积(5、10、15和20 mL)的乙腈,考察乙腈添加体积对上述兽药添加回收率的影响。
结果表明,在两种基质中,乙腈体积大于15 mL时的提取率趋于稳定,均能达到60%以上。考虑到样品中脂肪成分的影响,最终确定其加入量为15 mL。代表性的3种兽药阿莫西林(RT: 2.24 min)、双氟沙星(RT: 8.03 min)和吡喹酮(RT: 24.42 min)的保留时间范围完全涵盖了上述99兽药的洗脱时间。从图2可见,3种代表性兽药在牛奶和猪肉中的回收率均平稳地保持在70%以上。
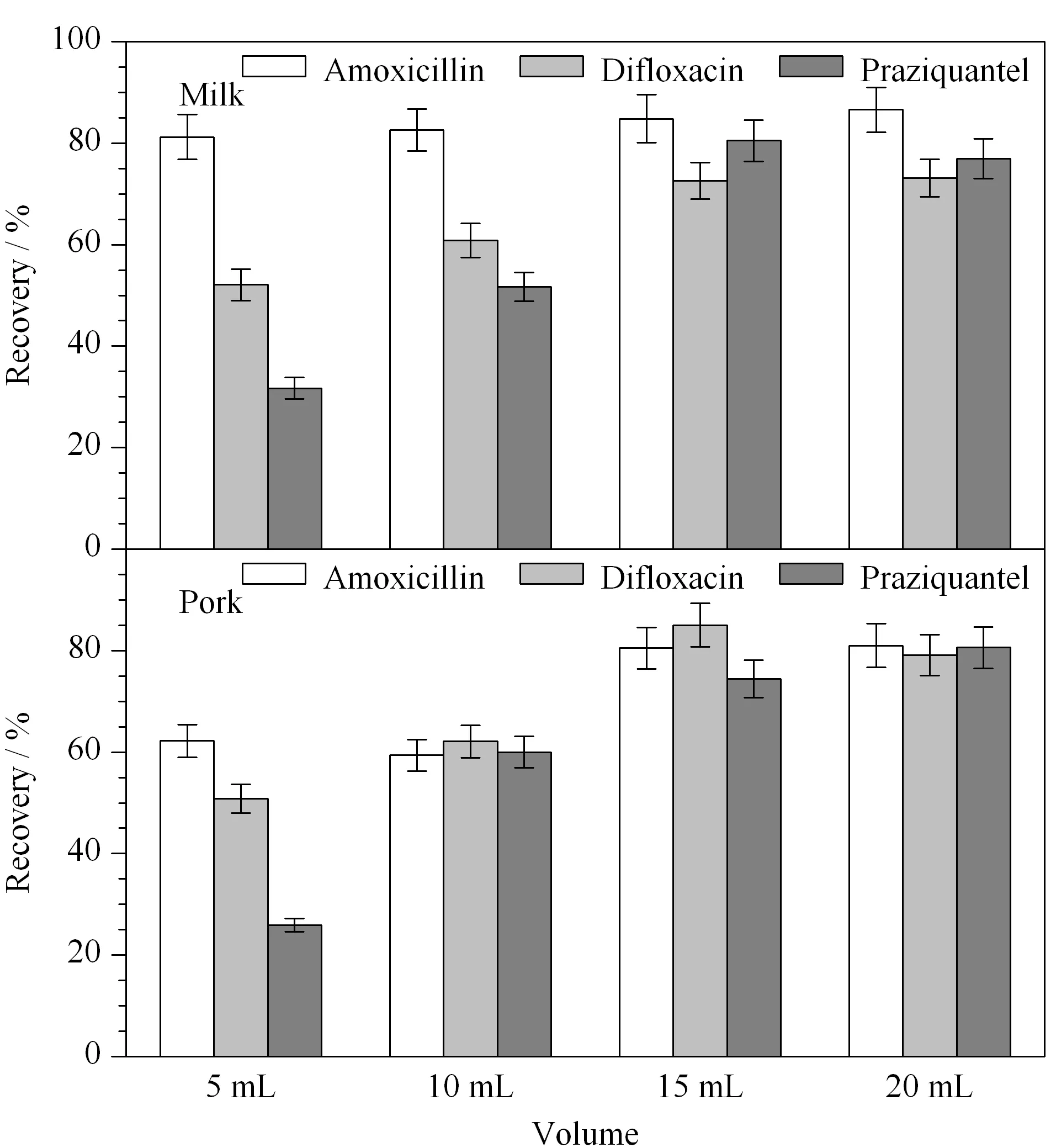
图 2 提取时乙腈体积对3种兽药加标回收率的影响(n=6)Fig. 2 Effect of the acetonitrile volume during extraction on the spiked recoveries of the three veterinary drugs (n=6)
2.4 洗脱体积的优化
洗脱液主要来自两部分,第一部分是作为辅助提取剂的乙腈,通过加入硫酸铵使乙腈从水相提取液中分离出来,得到的乙腈层位于离心管的上层,它作为水的辅助提取剂,很好地保证了弱极性兽药的回收率。水相提取液上样,位于离心管上层的乙腈作为洗脱液,逐级渗入,使一些强极性的兽药首先从饱和的硅藻土柱上洗脱下来。第二部分的洗脱液是纯乙腈,分别考查了10 mL、10 mL×2和10 mL×3乙腈作为洗脱溶剂对待测兽药回收率的影响。同样以代表性的3种兽药阿莫西林、双氟沙星和吡喹酮为例,从图3可见,当乙腈体积为10 mL×2和10 mL×3时,提取率趋于稳定,3种代表性兽药在牛奶和猪肉中的回收率均平稳地保持在70%以上。从节约氮吹浓缩的时间和成本的角度出发,确定纯乙腈作为洗脱液的洗脱体积为10 mL×2。
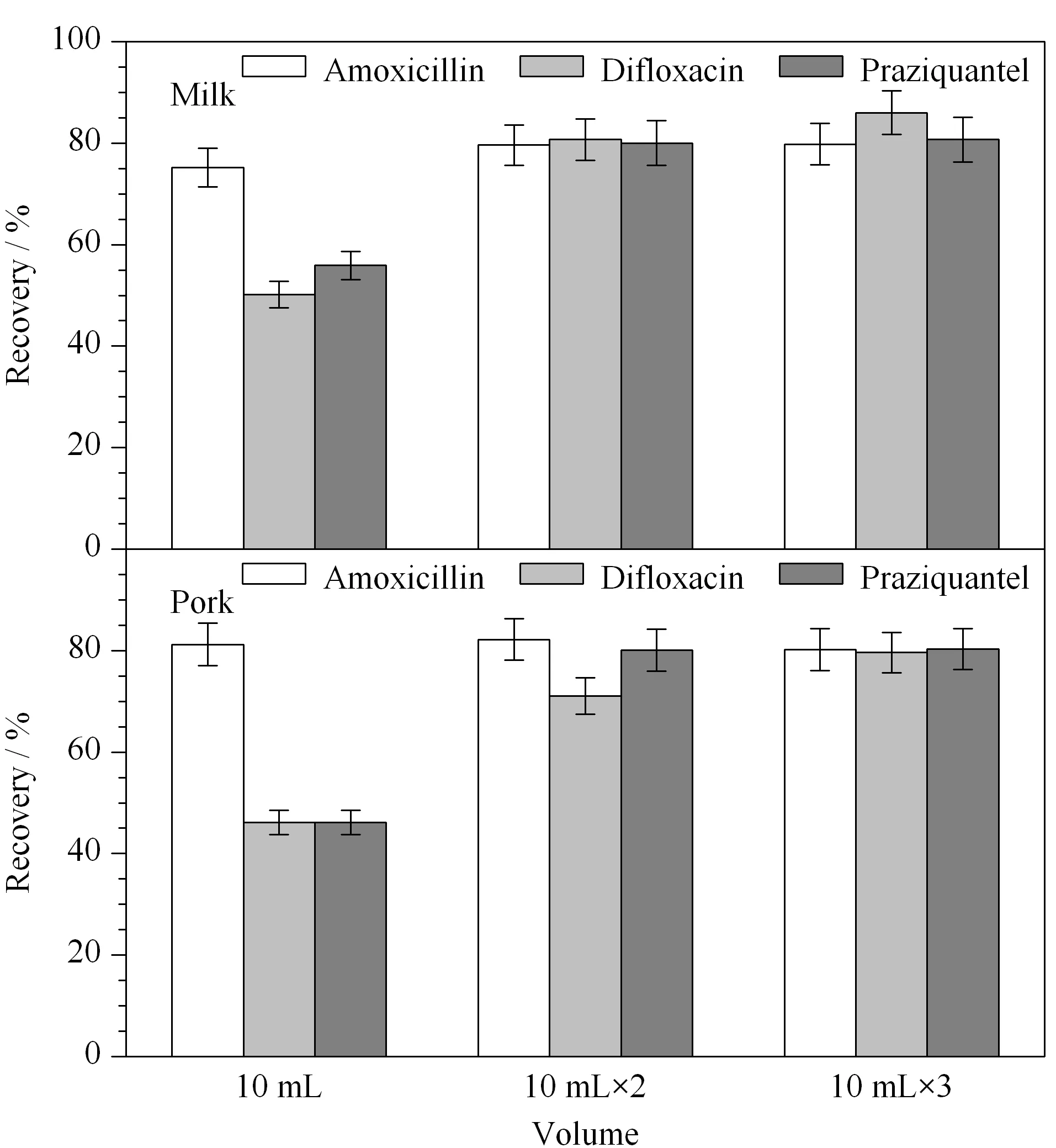
图 3 洗脱时乙腈体积对3种代表性兽药加标回收率的影响Fig. 3 Effect of the acetonitrile volume during elution on the spiked recoveries of the three veterinary drugs
2.5 液相色谱、质谱条件的优化
本文采用UHPLC联用高分辨台式Q-Exactive质谱,全扫描和正、负离子切换模式进行测定,在MS和MS/MS模式下完成正、负极性的快速切换,通过提取一级质谱的精确质量数进行定性和定量,自动触发二级,进一步提高定性的准确性,其超高的分辨率有助于解析复杂的样品,确保Q-Exactive系统能够在一次色谱运行中最大限度地检测和鉴定代谢物。
99种禁限用兽药种类和性质差别较大,为防止在混合标准溶液进样时不同性质化合物发生相互反应,根据各物质的性质将其分为3组:第一组为28种激素类化合物,第二组为39种磺胺喹诺酮类化合物,第三组为32种其他兽药。参照99种兽药的电离性质,通过流动注射泵连续进样,对每种兽药的单标溶液进行全扫描,确定每种兽药的电离方式和分子离子峰。其中部分激素类化合物采用ESI-模式,其余化合物采用ESI+模式。再分别输入各目标化合物的分子式,由软件计算精确分子式,可用工作站软件自动解析待分析样品的质谱扫描图谱,综合各种同位素丰度匹配比例、各种加合离子(如加H+、加Na+、加K+、二聚体、脱水峰、各种中性丢失),计算结果中精确质量数对应的可能的中性分子式组合,获得每个化合物的分子离子峰后,进行二级质谱扫描,优化碰撞能量,获得碎裂片段。与建立的目标数据库中化合物的分子式、碎裂片段质量数比较,并结合色谱保留时间,筛查是否包含数据库中兽药,从而实现目标兽药的快速筛查。
在流动相中加入0.1%(v/v)甲酸能增加正离子检测模式下兽药的电离效率,促进[M+H]+离子生成,因此针对正离子模式下检测的兽药,添加了0.1%(v/v)甲酸。同时也进一步考察了乙酸铵缓冲液离子强度的影响,乙酸铵溶液浓度从2 mmol/L变化到10 mmol/L的结果表明,以乙腈-5 mmol/L乙酸铵溶液(含0.1%(v/v)甲酸)作为流动相时获得了最优的色谱峰形、分离效果和质谱信号响应。相应地,负离子模式测定的激素类兽药则以含0.1%(v/v)氨水的水-乙腈溶液作为流动相。全过程选择梯度洗脱方式,通过优化流动相梯度洗脱条件实现了99种兽药的有效分离。
另外,比较了Hypersil Gold C18色谱柱和Hypersil Gold C8色谱柱,发现两柱只显示出组分保留时间的差异,在分离度方面均可满足质谱检测的需要。另外,针对2款不同规格的色谱柱(100 mm×2. 1 mm, 1.9 μm;100 mm×2.1 mm, 3 μm)进行了比较,同样压力下,结果也只显示出保留时间的极小差异。由于小粒径色谱柱流速更低,可以很好地节约有机溶剂,因此最终选择色谱柱为Hypersil Gold C18柱(100 mm×2.1 mm, 1.9 μm)。
2.6 加标回收试验结果的分析
各兽药的线性范围为0.001~0.1 μg/mL,检出限为0.000 5~0.02 mg/kg,加标回收率为60%~120%,标准偏差在15%以内(见表2~表8)。
结果表明,本方法对液态乳、猪肉、鱼类等食品基质具有较强的适用性,无论是脂溶性还是水溶性兽药,都可以一步完成提取、净化流程。
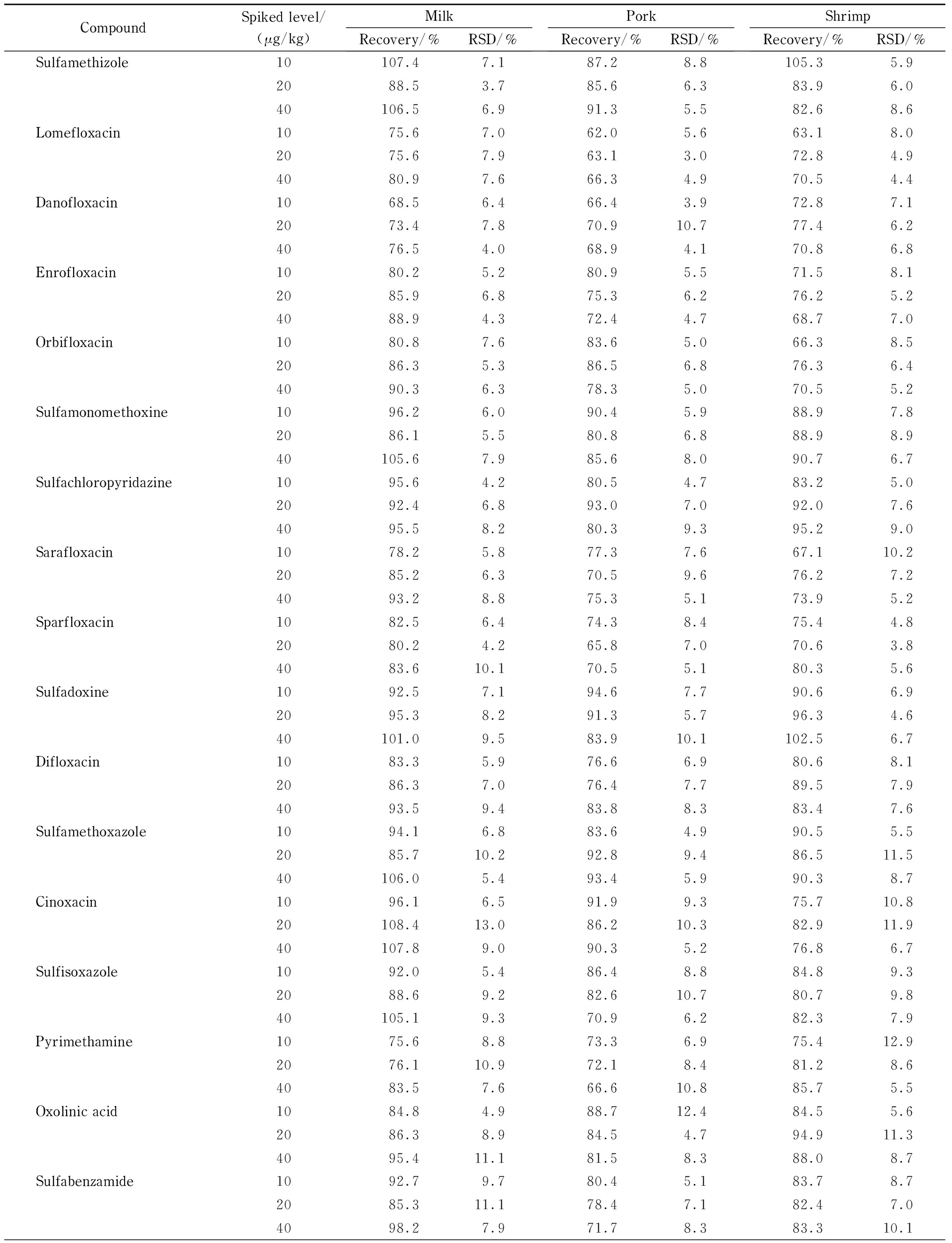
表 2 (续)
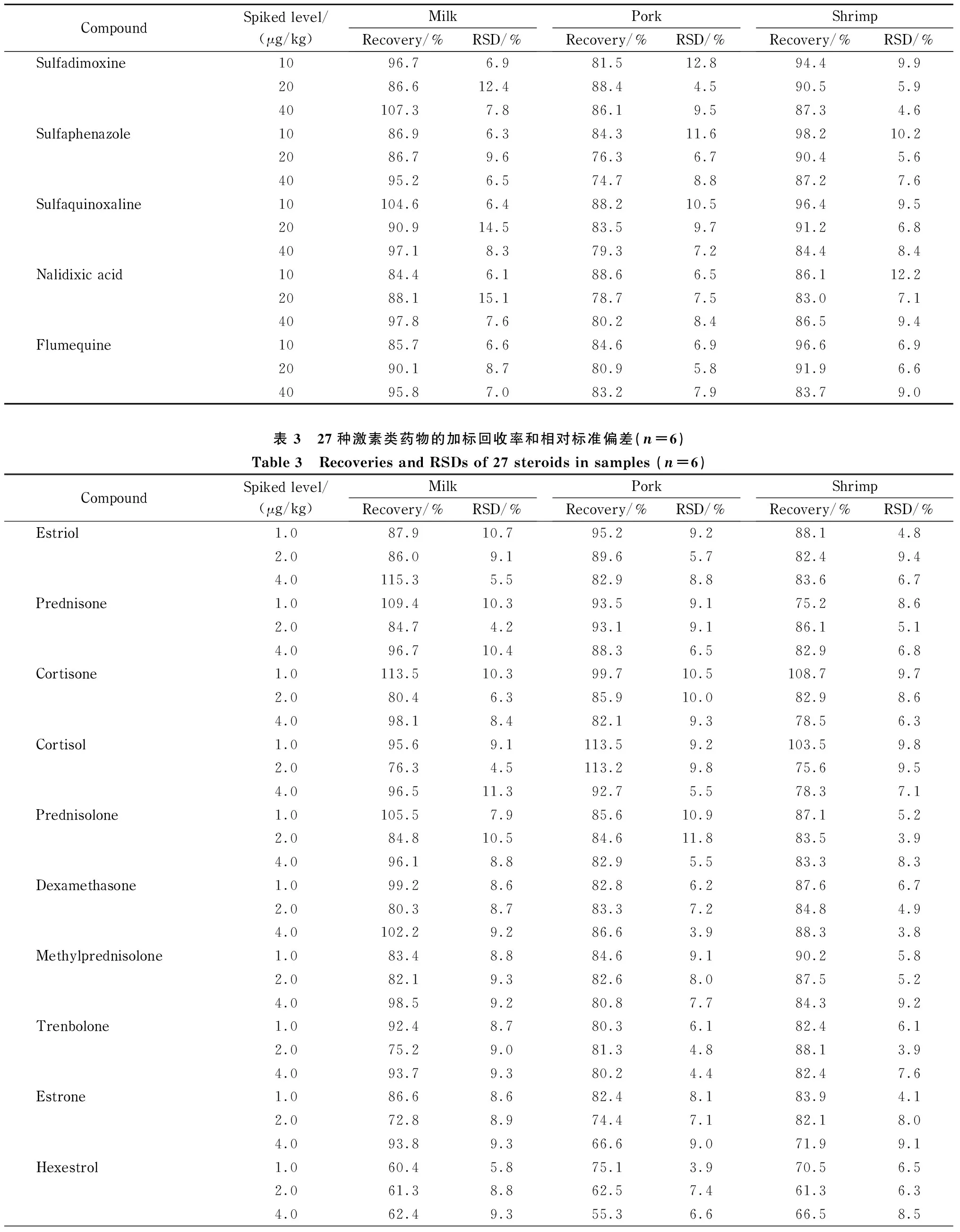
表 2 (续)
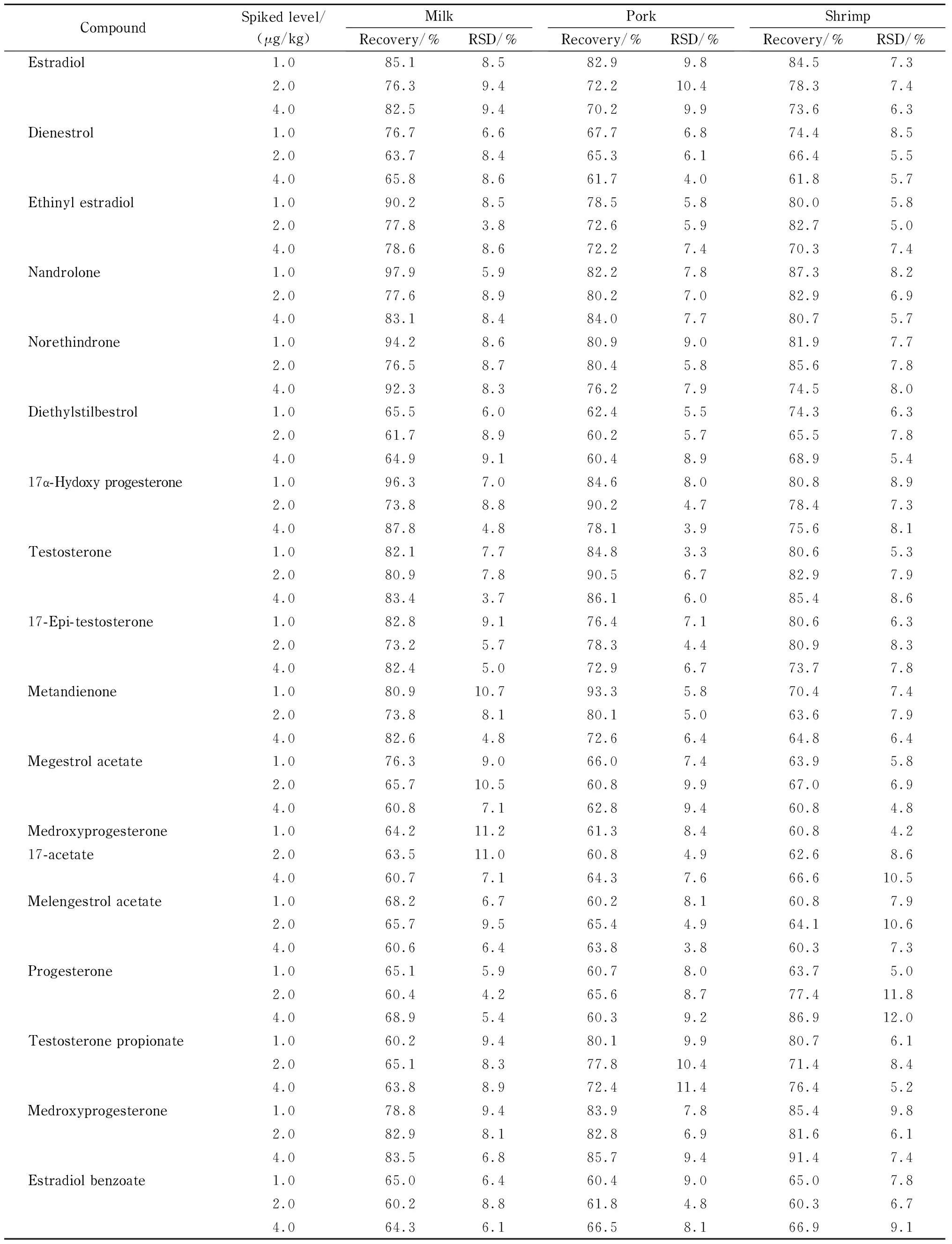
表 3 (续)
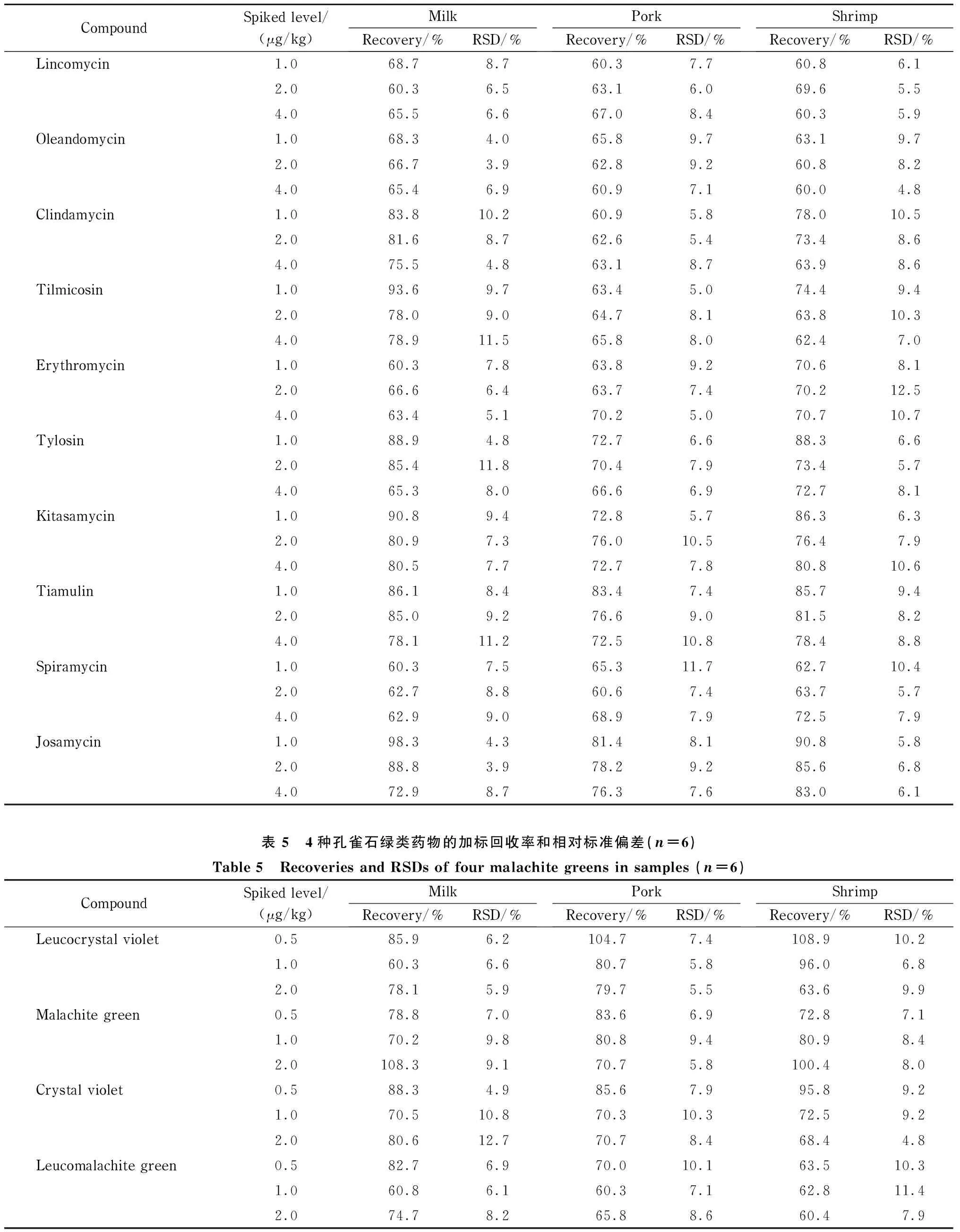
表 4 10种红霉素类药物的加标回收率和相对标准偏差(n=6)
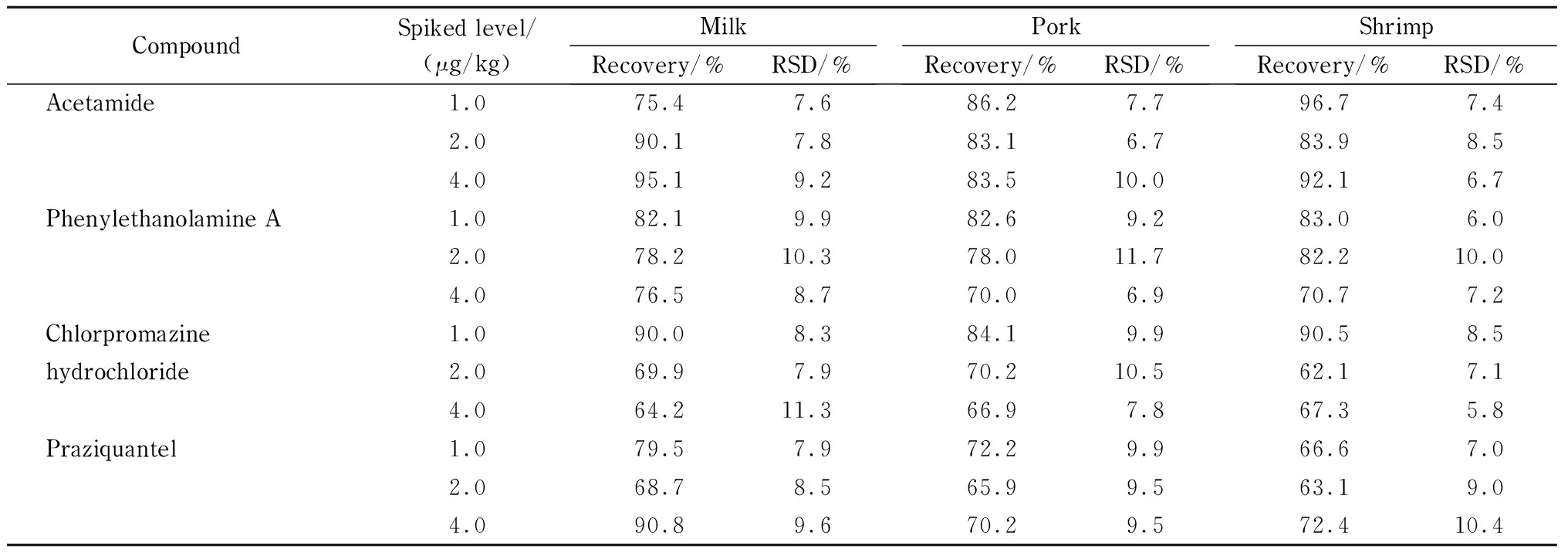
表 8 (续)
3 结论
基于载体辅助液液萃取技术,通过一次性前处理完理化性质差异很大的常见8大类共计99种兽药残留的提取、净化,同时结合高效液相色谱-四极杆/静电场轨道阱高分辨质谱进行测定,20 min内可完成99种兽药残留的高通量筛选和确认。高分辨率质谱在复杂基质样品分析中消除了基质的干扰,提高了定性和定量的准确性,自动触发模式提供的二级碎片进一步提高了定性的准确性。方法的检出限可满足国内外相关兽药残留限量的要求,可作为一种快速筛选和确证检测技术广泛使用。
参考文献:
[1] Li J H. Chinese Animal Husbandry and Veterinary Abstracts, 2018, 34(1): 9
李俊红. 中国畜牧兽医文摘, 2018, 34(1): 9
[2] Yang X L, Guo W W, Sun Q. Chinese Fishery Quality and Standards, 2013, 3(4): 1
杨先乐, 郭微微, 孙琪. 中国渔业质量与标准, 2013, 3(4): 1
[3] Yan H, Yun H, Liu X. Journal of Instrumental Analysis, 2013, 32(8): 909
严华, 云环, 刘鑫. 分析测试学报, 2013, 32(8): 909
[4] Meng Z, Shi Z H, Lü Y K, et al. Chinese Journal of Analytical Chemistry, 2014, 42(10): 1493
孟哲, 石志红, 吕运开, 等. 分析化学, 2014, 42(10): 1493
[5] Li F G, Su M, Li X Y. Chinese Journal of Chromatography, 2011, 29(2): 120
李锋格, 苏敏, 李晓岩. 色谱, 2011, 29(2): 120
[6] Wang L, Li Y Q, Wang H B. Chinese Journal of Analytical Chemistry, 2011, 39(2): 203
王炼, 黎源倩, 王海波. 分析化学, 2011, 39(2): 203
[7] Wang C Y, Wang Y P, Wang N. Chinese Journal of Analytical Chemistry, 2013, 41(1): 83
王重洋, 王远鹏, 王宁. 分析化学, 2013, 41(1): 83
[8] Xi J Z, Song J, Wang G N. Heilongjiang Animal Science and Veterinary Medicine, 2015(1): 53
锡建中, 宋娇, 王庚南. 黑龙江畜牧兽医, 2015(1): 53
[9] Chen S B, Shan Z J, Hu Q H. Food Science, 2004(12): 152
陈树兵, 单正军, 胡秋辉. 食品科学, 2004(12): 152
[10] Yu J. [MS Dissertation]. Hubei: Huazhong Agricultural University, 2012
于洁. [博士学位论文]. 湖北: 华中农业大学, 2012
[11] Zhang X G, Zheng Y M, Li Q, et al. Journal of the Hebei Academy of Sciences, 2014, 31(4): 59
张晓光, 郑雅梅, 李强, 等. 河北省科学院学报, 2014, 31(4): 59
[12] Qu B. Food Science, 2013, 34(5): 327
曲斌. 食品科学, 2013, 34(5): 327
[13] Quintana J B, Rodriguez I. Anal Bioanal Chem, 2006, 384: 1447
[14] Li X W, Guo P, Shan Y W, et al. J Chromatogr A, 2017, 1499(26): 57
[15] Gao F D, Zhao Y, Shao B, et al. Chinese Journal of Chromatography, 2012, 30(6): 560
高馥蝶, 赵妍, 邵兵, 等. 色谱, 2012, 30(6): 560
[16] Chu X G, Yong W, Ling Y, et al. Chinese Journal of Chromatography, 2007, 25(6): 907
储晓刚, 雍炜, 凌云, 等. 色谱, 2007, 25(6): 907
[17] Zhao Y S, Yang M L, Zhang F, et al. Chinese Journal of Chromatography, 2011, 29(7): 631
赵延胜, 杨敏莉, 张峰, 等. 色谱, 2011, 29(7): 631
[18] Didier O, Emmanuelle C, Philippe J, et al. J Chromatogr B, 2009, 877(23): 2363
[19] Chepyala D, Tsaide I L, Liao H W, et al. J Chromatogr A, 2017, 1491(31): 57
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23 06:22:32
煤化工(2022年3期)2022-07-08 07:24:42
中国粮油学报(2019年4期)2019-07-12 09:06:44
中成药(2018年2期)2018-05-09 07:20:06
中成药(2017年4期)2017-05-17 06:09:28
河南畜牧兽医(2016年24期)2016-11-29 01:28:18
中国资源综合利用(2016年10期)2016-01-22 08:36:09
兽医导刊(2015年7期)2016-01-04 11:59:54
西南军医(2015年1期)2015-01-22 09:08:16
云南畜牧兽医(2014年4期)2014-02-28 21:25:33
