
QuEChERS结合高效液相色谱串联质谱快速检测速溶茶粉中八种农药残留
2018-06-14 08:06鲍鏖天裴少芬唐杏燕王晓霞邵增琅
食品与发酵工业 2018年5期
鲍鏖天,裴少芬,唐杏燕,王晓霞,邵增琅
(南京融点食品科技有限公司,江苏 南京,211300)
速溶茶粉是以茶叶或茶鲜叶为主要原料,经过萃取、过滤、浓缩和干燥等工艺制成的茶叶深加工产品,由于具有冲调速溶、易与其他食品调配等特点,近年来取得了高速发展[1]。
温湿的茶园环境和较高的茶树种植密度使得茶树很容易滋生病虫害,现阶段以施用农药为主的防治措施又必然会带来农药残留问题[2]。以茶叶为原料生产速溶茶粉的过程中还会发生农药残留富集现象。以茶叶为参照,欧盟等主要进口国对速溶茶粉中的农药最高残留限量(maximum residue limit,MRL)要求非常严苛,我国最新修订的国标中也针对茶叶新增了多种农药检测项目[3-4]。但是目前针对速溶茶粉中农药残留检测的报道非常少。
为了保证速溶茶粉的质量安全和顺利出口,必须建立起检测速溶茶粉中农药残留的方法。速溶茶粉中含有源于茶叶的多种成分(茶多酚、咖啡碱等)和加工过程中添加的多种辅料(柠檬酸、D-异抗坏血酸钠等),基质成分相比茶叶更加复杂。液相色谱-串联质谱(liquid chromatography-tandem mass spectrometry,LC-MS/MS)的高选择性、高灵敏度等特点使其适于检测基质成分复杂样品中的痕量农药残留[5]。QuEChERS(quick, easy, cheap, effective, rugged, safe)方法是一种简单、快速的样品前处理技术,近年来被广泛应用于农产品的农药残留检测[6]。2017年6月开始实施的GB 2763—2016针对茶叶增加了多项农药残留检测项目,其中克百威、内吸磷、灭线磷、甲胺磷、辛硫磷、吡蚜酮、硫环磷、氯唑磷使用了LC-MS/MS方法进行检测[4]。本文采用QuEChERS方法对速溶茶粉进行前处理,探索建立适合同时检测速溶茶粉中这8种农药残留的HPLC-MS/MS方法,以期实现对速溶茶粉中这8种农药残留的准确、快速检测。
1 材料与方法
1.1 材料与试剂
NaCl、MgSO4(AR级),国药集团化学试剂有限公司;MgSO4、N-丙基乙二胺(PSA)、石墨化碳黑(GCB):上海安谱实验科技股份有限公司;乙腈、甲酸:LC-MS级,美国Sigma-Aldrich公司;尼龙有机相微孔滤膜:0.22 μm,德国C&B公司。
克百威(carbofuran)、内吸磷(demeton(o+s))、灭线磷(ethoprophos)、甲胺磷(methamidophos)、辛硫磷(phoxim)、吡蚜酮(pymetrozine)、硫环磷(phosfolan)和氯唑磷(isozofos)标准品:100 μg/mL,德国Dr.Ehrenstorfer GmbH公司。
1.2 仪器与设备
AL204电子分析天平,梅特勒-托利多仪器上海有限公司;XW-80A旋涡混合仪,金坛希望科研仪器有限公司;RJ-TGL-1650台式高速离心机,无锡瑞江分析仪器有限公司;明澈TM-D 24UV超纯水系统,美国Merck Millipore公司;1260-6420液相色谱串联质谱仪,美国Agilent公司。
1.3 实验方法
1.3.1 质谱条件
电喷雾离子源,正离子模式(ESI+);干燥气(氮气)流速和温度分别为10 L/min和350 ℃;雾化气压力为;毛细管电压为3 500 V。
1.3.2 液相色谱条件
色谱柱:Poroshell 120 EC-C18色谱柱(2.7 μm,3.0 mm×50 mm);柱温:40 ℃;进样量:5 μL;流速:0.3 mL/min。
流动相A:乙腈(含0.1%甲酸);流动相B:水(含0.1%甲酸)。
梯度洗脱程序:0~2 min,10% A;2~3 min,10%~60% A;3~10 min,60% A;10~11 min,60%~90% A;11~15 min,90% A;15~16 min,90%~10% A;16~20 min,10% A。
1.3.3 样品前处理
称取1 g(精确至0.01 g)样品于50 mL离心管中,加入5 mL去离子水和10 mL乙腈,旋涡振荡3 min,加入2.5 g NaCl,旋涡振荡2 min,10 000 r/min离心5 min。
移取1 000 μL上层乙腈提取液加入到装有150 mg MgSO4、50 mg PSA和50 mg GCB的2 mL离心管中,旋涡振荡1 min,15 000 r/min离心5 min,移取200 μL净化液至2 mL离心管中,加入800 μL超纯水,旋涡振荡1 min后过微孔滤膜备用。
1.3.4 溶液配制
混合标准储备溶液:分别移取100 μL 100 μg/mL农药标准溶液于10 mL容量瓶中,用乙腈定容,配制成1 μg/mL混合标准储备溶液。4 ℃下避光保存。
混合标准溶液:移取100 μL混合标准储备溶液于2 mL的离心管中,加入900 μL超纯水,配制成100 ng/mL混合标准溶液;移取100 μL 100 ng/mL混合标准溶液于2 mL离心管中,加入900 μL超纯水,配制成10 ng/mL混合标准溶液。混合标准溶液现配现用。
溶剂标准工作液:分别移取200 μL乙腈于2 mL离心管中,再移取相应浓度和体积的混合标准溶液于各离心管中,加水至1 mL,旋涡振荡1 min后过微孔滤膜,配制成0.2、0.5、1、5、10和50 ng/mL溶剂标准工作液。
基质匹配标准工作液:分别移取200 μL空白基质净化液(按1.3.3提取、净化)于2 mL离心管中,再移取相应浓度和体积的混合标准溶液于各离心管中,加水至1 mL,旋涡振荡1 min后过微孔滤膜,配制成0.2、0.5、1、5、10和50 ng/mL基质匹配标准工作液。
2 结果与分析
2.1 质谱条件的优化
ESI+模式下,用全扫描、单离子监测扫描和子离子扫描优化各化合物母离子的碎裂电压(fragmentor)和子离子的碰撞能(collision energy,CE)。空白基质进样,多重反应监测模式(multiple reaction monitoring,MRM)下发现,在灭线磷243.1>173.0的离子通道上存在基质干扰,在其他离子通道上均没有基质干扰。参考BOTERO-COY等检测土壤中草甘膦时遇到基质干扰时的试验方法,虽然子离子173.0的响应高于96.9的响应,但选择没有基质干扰的243.1>96.9作为灭线磷的定性离子对[7]。其他7种化合物均选择响应最高的2个子离子作为定量和定性离子。优化后的质谱MRM参数见表1。
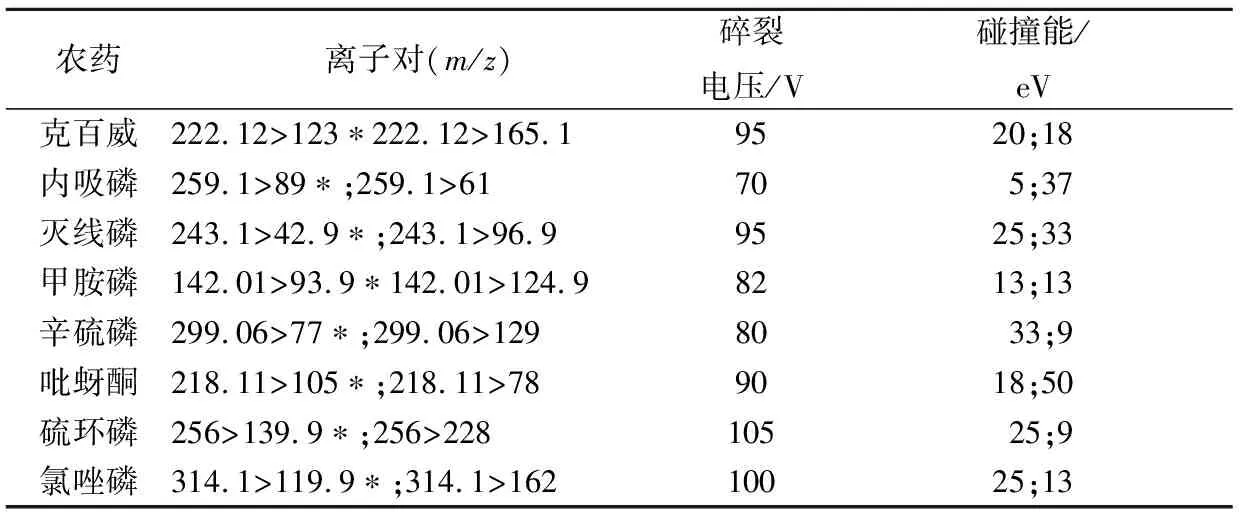
表1 8种农药的MRM参数Table 1 The MRM parameters of the 8 kinds of pesticides
注:*为定量离子。
2.2 液相色谱条件的优化
2.2.1 流动相的选择
为了减小溶剂效应,选择与萃取溶剂相同的乙腈作为有机相[8]。ESI+模式下,在LC-MS/MS的流动相加入挥发性的酸可以提供H+,从而提高目标化合物的离子化效率[6]。综合考虑色谱柱的pH耐受范围和目标化合物的离子化效率,分别在有机相和水相中加入0.1%的甲酸。
2.2.2 洗脱梯度的优化
吡蚜酮的提取离子流图(extracted ion current,EIC)如图1所示。在1~2 min有机相比例为5%的条件下,吡蚜酮的峰型发生裂分,表现出很强的溶剂效应[8-9]。将1~2 min有机相比例提高到10%,吡蚜酮峰型裂分现象消失,峰型良好。8种农药的总离子流图(total ion current,TIC)如图2所示。由图2可知,2~3 min有机相比例快速提高到60%后并维持7 min,8种农药在10 min内全部出峰且基本分离开。质谱检测与色谱检测不同,不需要通过色谱将各目标化合物完全分离,但保证目标化合物具备一定分离度可以减小基质与目标化合物共流出的概率,从而减小目标化合物离子化时的基质干扰[10]。
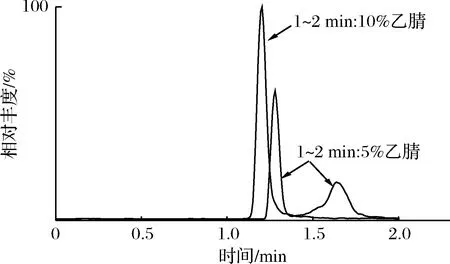
图1 吡蚜酮的提取离子流图Fig.1 The EIC of the pymetrozine

图2 8种农药的总离子流图Fig.2 The TIC of the 8 kinds of pesticides
2.3 前处理条件的优化
2.3.1 盐的选择
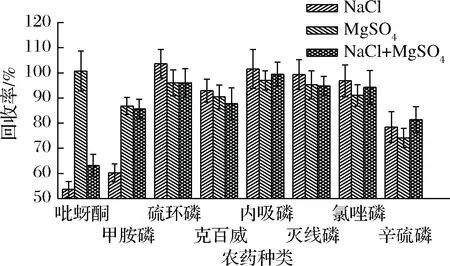
图3 不同盐对农药回收率的影响Fig.3 The influence of different salt on recoveries of pesticides
2.3.2 净化剂的选择
PSA可以显著吸附茶叶中的茶多酚和叶绿素,GCB可以显著吸附茶叶中的咖啡碱并净化提取液的颜色[13]。在1.3.3的样品前处理过程中分别加入150 mg MgSO4+50 mg PSA和150 mg MgSO4+50 mg PSA+50 mg GCB,试验重复3次[6]。各农药回收率试验结果如图4所示。以MgSO4+PSA+GCB作为净化剂时,吡蚜酮和辛硫磷的平均回收率分别只有37.7%和54.1%,而以MgSO4+PSA作净化剂时,2种化合物的平均回收率分别为108.5%和92.3%。这可能是因为吡蚜酮和辛硫磷的分子结构的平面化程度比较高(2种化合物均含有环状平面结构),净化过程中对平面化合物具有吸附作用的平面层状结构的GCB吸附了这2种化合物[6]。综合考虑8种农药的回收率,本文选择使用MgSO4+PSA。
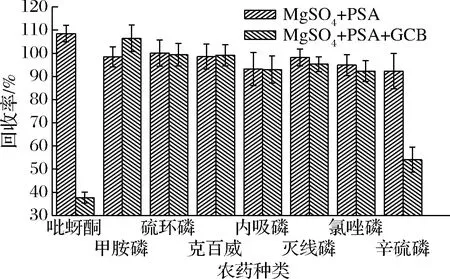
图4 不同净化剂对农药回收率的影响Fig.4 The influence of different adsorbent on recoveries of pesticides
2.4 基质效应
基质效应(matrix effect,ME)严重影响LC-MS/MS分析的准确性。KMELLR等用基质匹配标准曲线的斜率和溶剂标准曲线的斜率的差异(%)来衡量基质效应的强弱,ME>20%表明有比较强的基质增强效应,ME<-20%表明有比较强的基质抑制效应[14]。
虽然本文通过净化提取液、色谱分离和基质稀释等方法减弱了基质效应,但如表2所示,样品中的吡蚜酮和甲胺磷仍然表现出较强的基质抑制效应。反相色谱分离时最初流出的主要是基质中的极性成分,可能这些成分大幅降低了吡蚜酮和甲胺磷在ESI源中的离子化效率,造成了较强的基质抑制效应[15-16]。本文利用基质匹配标准曲线来消除基质效应对定量分析的影响,以获得更准确的定量结果。
2.5 回收率和精密度
各农药的基质匹配标准曲线方程如表2所示。在LOQ~50 ng/mL范围内,8种农药的线性关系良好(R2均大于0.996)。
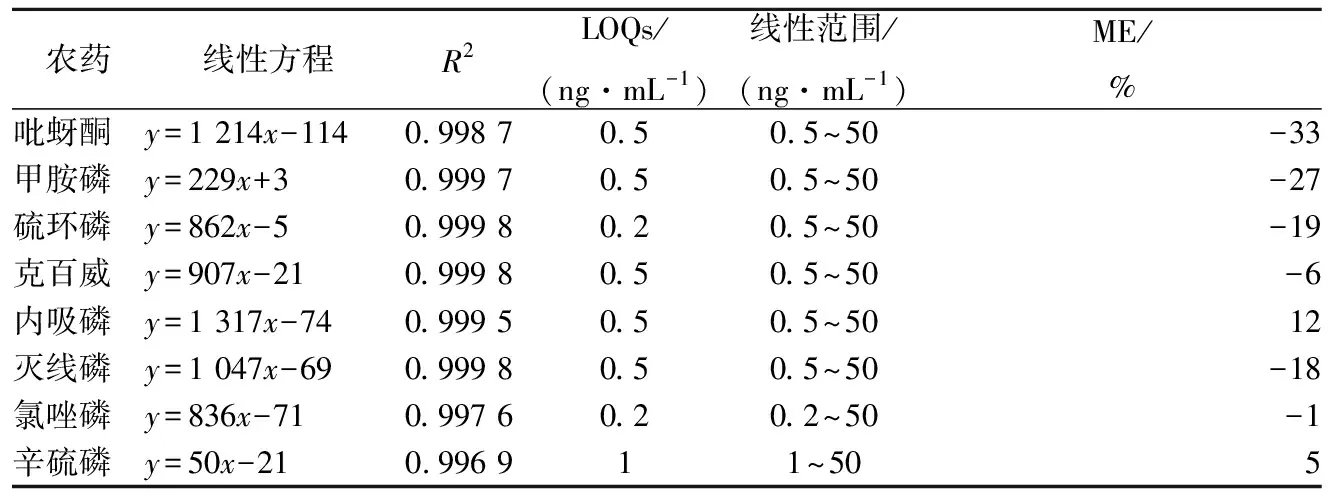
表2 农药的线性方程、相关系数(R2)、LOQs、线性范围和METable 2 The linear equations, coefficients of determination (R2), LOQs, linear ranges and ME obtained for pesticides
在1 LOQ和4 LOQ两个浓度添加水平下进行8种农药的回收率和精密度试验,试验重复6次[17]。回收率和精密度试验结果如表3所示。在2个浓度添加水平下吡蚜酮的平均回收率分别为61%和63%,回收率偏低。这可能是因为速溶茶粉中的一些成分对吡蚜酮有吸附作用,造成乙腈萃取吡蚜酮的效率偏低。8种农药在2个浓度添加水平下的平均回收率均在61%~118%,RSD均在0.9%~10.5%,符合美国分析家化学协会(AOAC)对于农产品中农药残留检测分析的要求(回收率在50%~150%,RSD<15%)[6]。本方法的方法定量限小于或等于国标和欧盟法令对茶叶及相关制品中这8种农药最大残留限量的要求[18-21]。本方法可以满足对速溶茶粉中这8种农药残留检测的要求。
2.6 实际样品的测定
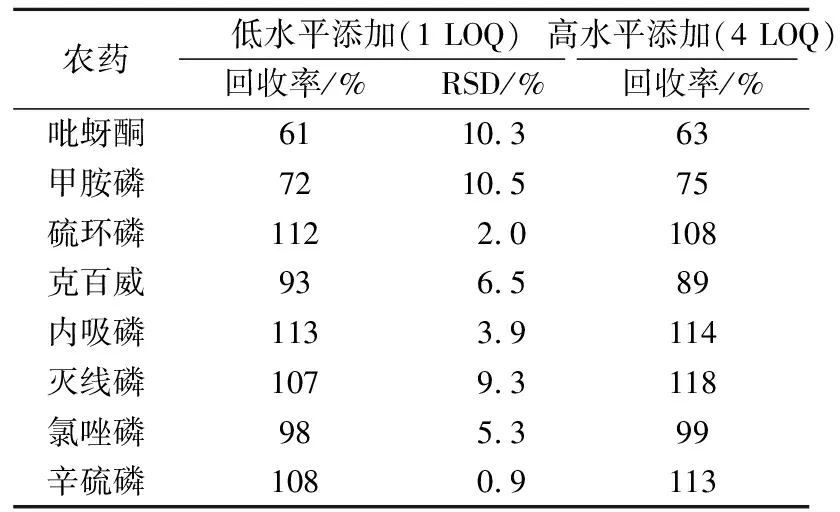
表3 农药的回收率和精密度Table 3 The recoveries and RSDs of pesticides spiked atlow and high concentrations
应用本方法检测10个批次用低农药残留茶叶生产的速溶茶粉和10个批次用普通茶叶生产的速溶茶粉。在10个批次的低农速溶茶粉中均没有检出这8种农药残留。在1个批次的普通速溶茶粉中检出了0.032 mg/kg的甲胺磷残留,虽然低于国标和欧盟法令的要求(0.05 mg/kg和0.1 mg/kg),但速溶茶粉中检出高毒的禁用农药仍应引起足够的重视。
3 结论
本文研究建立了QuEChERS前处理结合HPLC-MS/MS技术快速检测速溶茶粉中8种农药残留的方法,系统优化了方法中的质谱、色谱和前处理条件。良好的线性关系、回收率和精密度等指标表明本方法可以对速溶茶粉中的这8种农药残留进行准确的定量检测。同时本方法灵敏度高、快速、消耗的有机溶剂少,以本方法为基础可以进一步开发速溶茶粉中其他多种农药残留快速检测的方法。
[1] 邹锋扬, 金心怡, 王淑凤, 等.速溶茶粉产品的研究进展[J].饮料工业, 2012, 15(3):7-12.
[2] 冯洁.茶叶中农药残留分析方法的应用研究[D].北京:北京化工大学, 2014.
[3] 陈宗懋.2008年欧盟发布茶叶中农药残留新标准[J].中国茶叶, 2008, 30(4):7-7.
[4] 中华人民共和国国家卫生和计划生育委员会, 中华人民共和国农业部, 国家食品药品监督管理总局.GB 2763—2016食品国家安全标准 食品中农药最大残留限量[S].北京: 中国标准出版社, 2016.
[5] 盛龙生.色谱质谱联用技术[M].北京:化学工业出版社, 2006.
[6] ANASTASSIADES M, LEHOTAY S J,TAJNBAHER D, et al.Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce[J].Journal of AOAC International, 2003, 86(2): 412-431.
[7] BOTERO-COY A M, IBEZ M, SANCHO J V, et al.Improvements in the analytical methodology for the residue determination of the herbicide glyphosate in soils by liquid chromatography coupled to mass spectrometry[J].Journal of Chromatography A, 2013, 1 292: 132-141.
[8] 王丽, 申兰慧, 陈国清.浅谈样品溶剂对高效液相色谱行为的影响[J].中国药事, 2013, 27(2):163-166.
[9] WILLIAMS K J, PO A L W, IRWIN W J.Sample-solvent-induced peak broadening in the reversed-phase high-performance liquid chromatography of aspirin and related analgesics[J].Journal of Chromatography A, 1980, 194(2): 217-223.
[10] CHAMBERS E, WAGROWSKI-DIEHL D M, LU Z, et al.Systematic and comprehensive strategy for reducing matrix effects in LC/MS/MS analyses[J].Journal of Chromatography B, 2007, 852(1): 22-34.
[12] 黎国富, 杨劲, 赵浩如.盐析辅助均相液液萃取法在体内药物分析中的应用进展[J].药学进展, 2010, 34(7):313-318.
[13] 尹鹏, 陈红平, 刘新, 等.5种分散吸附剂对茶叶乙腈提取液组分的吸附作用研究[J].分析试验室, 2013, 32(6): 54-58.
[15] NIESSEN W M A, MANINI P, ANDREOLI R.Matrix effects in quantitative pesticide analysis using liquid chromatography-mass spectrometry[J].Mass Spectrometry Reviews, 2006, 25(6): 881-899.
[16] VAN EECKHAUT A, LANCKMANS K, SARRE S, et al.Validation of bioanalytical LC-MS/MS assays: evaluation of matrix effects[J].Journal of Chromatography B, 2009, 877(23): 2 198-2 207.
[17] 中华人民共和国国家卫生和计划生育委员会, 中华人民共和国农业部, 国家食品药品监督管理总局.GB 23200.13—2016食品国家安全标准 茶叶中448种农药及相关化学品残留量的测定 液相色谱-质谱法[S].北京: 中国标准出版社, 2016.
[18] Commission Decision 76/895/EEC[S].Official Journal of the European Communities, 2007.
[19] Commission Decision 86/362/EEC[S].Official Journal of the European Communities, 2013.
[20] Commission Decision 86/363/EEC[S].Official Journal of the European Communities, 2013.
[21] Commission Decision 90/642/EEC[S].Official Journal of the European Communities, 2013.
猜你喜欢
煤化工(2022年3期)2022-07-08
中国土壤与肥料(2021年5期)2021-12-02
色谱(2021年7期)2021-06-07
今日农业(2020年22期)2020-12-14
科技视界(2020年26期)2020-09-24
科技视界(2020年17期)2020-07-30
化学教学(2018年1期)2018-02-28
中国资源综合利用(2016年10期)2016-01-22
电力需求侧管理(2014年4期)2014-03-20
中国药房(2012年10期)2012-08-07
