
Toll样受体4介导的巨噬细胞泡沫化与动脉粥样硬化*
2018-06-14 02:35高丽娜
济宁医学院学报 2018年3期
高丽娜
(济宁医学院药学院,日照 276826)
动脉粥样硬化为主要特征的血管性疾病正迅速成为全球性死亡的主导原因,占总死亡率20%。针对动脉粥样硬化病理机制,研究者先后提出了“脂质浸润学说”、“动脉平滑肌细胞增殖学说”、“血栓源性学说”、“损伤反应学说”和“炎症学说”等,目前动脉粥样硬化发病机制仍不完全明确[1]。动脉粥样硬化是由多种因素引起的复杂性疾病,目前,公认的发病机制为“慢性血管炎性学说”[2]。动脉粥样硬化斑块的形成始于血源性炎细胞在脂质沉积或动脉损伤位点的募集,动脉粥样硬化每一阶段都有细胞因子和趋化因子的参与。巨噬细胞是动脉粥样硬化的重要参与者,对其发病机制起决定性作用[3-4]。Toll样受体(TLRs)是一组模式识别受体,可识别各种微生物产物,与配体结合后,激活机体的免疫反应。同时,TLRs也可被内源性配体激活,如坏死细胞和修饰的脂蛋白成分[5]。在人或者啮齿动物动脉粥样硬化中,巨噬细胞表面均有TLR4表达。本文从炎症反应和脂质代谢紊乱两个方面对巨噬细胞表面TLR4活化在动脉粥样硬化发病机制中的作用作一综述。
1 TLR4介导巨噬细胞炎症反应在动脉粥样硬化中的作用
动脉粥样硬化是一种慢性血管炎性疾病,参与动脉粥样硬化的细胞包括血管平滑肌细胞、内皮细胞、单核细胞、巨噬细胞等[6]。TLRs可见于不同类型的细胞中,如TLR2和TLR4在单核细胞、巨噬细胞、泡沫细胞、髓样树突状细胞、平滑肌细胞和B淋巴细胞中均有表达。人动脉粥样硬化的特征在于,TLR1、TLR2和TLR4主要在巨噬细胞和内皮细胞中表达量增加;而在鼠动脉粥样硬化中,TLR4只在巨噬细胞中表达量增加[4]。
巨噬细胞参与动脉粥样硬化发生和发展全过程,研究者采用激光捕获显微切割技术从损伤的巨噬细胞中分离和鉴定了炎症标志物,如TLR1、TLR2和TLR4[7]。耿红莲[8]研究发现,冠心病患者血清中抗氧化低密度脂蛋白(ox-LDL)抗体阳性率显著高于健康对照组,而且,外周血单核细胞中TLR4表达上调。进一步采用ox-LDL诱导人单核细胞THP-1,可引起TLR4表达上调,激活NF-κB,诱导单核细胞趋化因子(MCP-1)和IL-8分泌增加,而采用siRNA抑制TLR4表达后,上述炎症反应减弱。血凝素样氧化低密度脂蛋白受体(LOX)-1是ox-LDL的特异性受体,在动脉粥样硬化的炎症反应过程中,TLR4与LOX-1具有协同作用,相互激活。张颖[9]采用ox-LDL诱导人单核细胞THP-1从细胞水平模拟动脉粥样硬化,发现TLR4与LOX-1通过NF-κB途径诱导炎症介质IL-6、IL-12和TNF-α过量表达,参与并加剧动脉粥样硬化的炎症损伤过程。
Ox-LDL启动炎症反应主要通过活化TLR4/TLR6异源二聚体及其配体脂肪酸转移酶(CD36)发挥作用。然而,LDL中的脂质成分——人氧化磷脂(oxPAPC)可通过TLR4诱导IL-8转录增加,以一种非依赖CD14和CD36的方式促进炎症反应的发生[10]。TLR4活化可分别通过髓样分化因子(MyD)88依赖性和非依赖性(TIR结构域衔接蛋白,TRIF)途径启动下游信号转导调节转录因子NF-κB、c-Jun氨基末端激酶(JNK)和干扰素调节因子(IRF)的活性,促进炎症因子和干扰素(IFN)β的过量表达。MyD88基因敲除(MyD88-/-)后,小鼠动脉粥样硬化样症状得到明显改善[11]。在人动脉粥样硬化的研究中,抑制MyD88基因表达后,其下游的NF-κB/AP-1转录因子表达也受到抑制,进而导致炎症因子和MMP-9等含量下降[12]。因此,在动脉粥样硬化巨噬细胞泡沫化过程中,ox-LDL活化TLR4并通过MyD88和TRIF启动下游的NF-κB/AP-1/IRF,分泌大量的炎症因子、趋化因子等介导炎症反应,是脉粥样硬化主要的损伤机制之一。见图1。
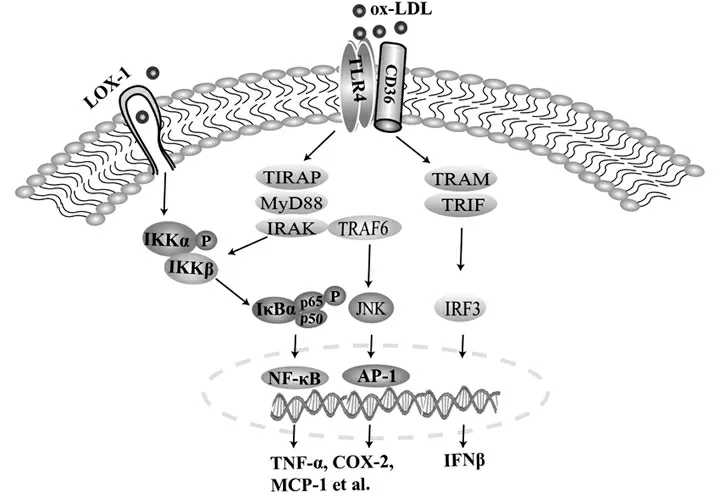
注:LOX-1,血凝素样氧化低密度脂蛋白受体;ox-LDL,氧化低密度脂蛋白;CD36,脂肪酸转移酶;TLR4,Toll样受体4;TIRAP,Toll/白介素-1受体相关蛋白;MyD88,髓样分化因子88;IRAK,白介素-1受体相关激酶;IKK,IκB激酶;IκB,NF-κB,抑制剂;NF-κB,核因子κB;AP,转录因子活化蛋白;TRAF,肿瘤坏死因子受体相关因子;JNK,c-Jun氨基末端激酶;TRAM,Toll样受体相关分子;TRIF,TLR4信号通路干扰素TIR结构域衔接蛋白;IRF3,干扰素调节因子3;TNF-α,肿瘤坏死因子α;COX-2,环氧合酶2;MCP-1:单核细胞趋化因子1;IFNβ:β干扰素
图1 ox-LDL活化TLR4介导的巨噬细胞炎症反应机制模式图
2 TLR4调控巨噬细胞脂质谢紊乱在动脉粥样硬化中的作用
脂质代谢异常与免疫反应紧密相关。血浆胆固醇和甘油三酯升高的小鼠对细菌或病毒的易感性更强,这种血脂升高或血脂异常与TLRs活性有关[13]。
2.1 TLRs结构特点
TLRs是一型跨膜蛋白,由胞外区、跨膜区以及胞内区3部分组成,其胞浆结构域与IL-1受体家族高度相似,故被称为Toll/IL-1受体(TIR)结构域,具有高度保守性。Kberlin等[13]对TLRs的TIR和TMD结构域及其与局部脂质环境的相互作用进行了综述。脂质筏是细胞膜上富含胆固醇的区域,参与调节跨膜蛋白与膜微区的动态结合,并介导TLRs在细胞膜上的重定位[14]。TLRs的TMD结构域对同型二聚体或异型二聚体受体复合物的形成起重要作用,并介导受体分选与分子伴侣的相互作用。TLRs还参与和阴离子甘油磷脂如磷脂酰丝氨酸(PS)以及磷脂酰肌醇(PI)的相互作用[15]。TLR4有两个桥接因子:TIRAP/MAL和TRAM/TICAM2,前者是TLR4从细胞表面发出信号所必需的;后者则对内涵体的TLR4信号传导非常重要。虽然TLR4结构域及其与局部脂质环境的相互作用机制仍不清楚,但TLR辅助因子的特异性脂质缔合以及TLR4本身作为跨膜蛋白的特性,表明TLR4信号转导对局部膜环境具有较强的依赖性。TLR4在激活时被动态地募集到脂质筏中,说明细胞膜局部的胆固醇和鞘脂等会促进TLR4激活。见图2。

注:TMD,跨膜结构域;TIR,TLR结构域;PIP2,磷脂酰肌醇二磷酸结合结构域;G,甘氨酸;C,半胱氨酸
图2 TLRs与相关蛋白的膜相互作用介导脂质代谢和炎症反应[13]
2.2 TLR4与ABCA1、ABCG1
动脉粥样硬化的主要特征为ox-LDL附着于血管内皮下,被巨噬细胞吞噬后,巨噬细胞内胆固醇转运障碍引起巨噬细胞泡沫化,进而形成动脉粥样硬化斑块。巨噬细胞表面广泛地分布着介导修饰性LDL(如ox-LDL)摄入的受体,包括TLR2、TLR4、TLR6、清道夫受体(SR)A、CD36等[4]。ox-LDL/TLR4通过上调CD36介导巨噬细胞摄入大量ox-LDL,并触发炎症信号转导通路。调节细胞内胆固醇合成、吸收和外排是维持体内胆固醇平衡的必要生理过程。在细胞内,ox-LDL衍生的胆固醇酯进入溶酶体,经中性胆固醇酯酶(CEH)水解为游离胆固醇和脂肪酸[16]。游离胆固醇在细胞内存在多种代谢途径,膜转运蛋白——三磷酸腺苷结合盒转运体(ABC)A1、ABCG1是其主要代谢途径。ABCA1是影响高密度脂蛋白(HDL)生物合成和胆固醇逆向转运的关键因子。巨噬细胞过度表达ABCA1可促进巨噬细胞内胆固醇逆向转运并缓解动脉粥样硬化的症状[17]。此外,ABCA1直接与细胞外受体ApoA-I相连,促进胆固醇、鞘磷脂、磷脂酰胆碱等转移至细胞外[18-20]。ABCA1介导胆固醇和磷脂等向ApoA-I外排,使ApoA-I转化为新的HDL颗粒,而新生HDL是ABCG1介导胆固醇外排的受体,因此,ABCG1被认为在ABCA1的下游发挥作用,二者协同促进巨噬细胞内胆固醇流出,对动脉粥样硬化的发生和发展具有保护作用,但其具体机制仍不明确[21]。此外,过量的游离胆固醇可被转运到内质网,经过胆固醇酰基转移酶(ACAT)再次酯化为胆固醇脂肪酸酯(泡沫细胞的“泡沫”),作为脂滴存储于巨噬细胞内。
ABCA1基因突变引起ABCA1蛋白功能障碍,表现为血浆中HDL和ApoA1含量降低,细胞内胆固醇流出减少,但ABCA1基因突变是否为冠心病动脉粥样硬化的危险因素有待进一步研究[22]。除细胞内脂滴蓄积以外,细胞外胆固醇蓄积也可能加剧动脉粥样硬化的发展。因此,以ABCA1为靶点开发抗动脉粥样硬化药物应该兼顾ABCA1介导的胆固醇转运和外排作用。
ABCA1和ABCG1均能显著影响巨噬细胞的炎症免疫反应[23],可能与胆固醇蓄积导致膜脂质组成改变有关,进而影响TLRs的生物学作用。与野生型小鼠相比,ABCA1-/-小鼠巨噬细胞对TLR4激动剂诱导的炎症反应的响应性增强。从ABCA1-/-小鼠巨噬细胞脂质筏中分离胆固醇发现,与野生型小鼠比较,从ABCA1-/-小鼠巨噬细胞中胆固醇含量增加23%,细胞膜脂质筏中的TLR4含量也显著增加[24],提示ABCA1可能通过调节脂质筏中胆固醇含量以及转运至脂质筏的TLR4含量来调节巨噬细胞对TLR4激动剂的响应性。正常状态下,巨噬细胞ABCA1选择性地减少脂质筏中的游离胆固醇含量,并减少MyD88依赖性TLRs向脂质筏转运,抑制TLRs活化,进而降低炎性损伤。Ma等[25]研究发现ABCA1通过活化PKA将巨噬细胞诱导为M2型,促进抗炎细胞因子IL-10表达,从而发挥抗炎作用。上述研究提示,ABCA1介导的胆固醇转运影响了细胞膜脂质筏稳态,并通过TLR4与巨噬细胞活化相关联,但ABCA1与巨噬细胞炎症因子分泌的直接作用关系报道较少。同样地,TLR2和TLR4激活能调节小鼠单核/巨噬细胞株RAW 264.7细胞中ABCA1和ABCG1 mRNA的表达,涉及的下游信号转导通路包括NF-κB和p38MAPK[26]。但是,由于信号转导通路间复杂的网络交互性,TLRs与ABCA1和ABCG1之间存在复杂的关系,例如,TLR2和TLR4活化可引起ABCA1 mRNA表达上调,而对蛋白表达无显著性影响;TLR2和TLR4活化可引起ABCG1 mRNA表达下调,而蛋白表达上调等[22]。因此,TLR4、ABCA1、ABCG1与巨噬细胞炎症反应以及脂质代谢之间的相互作用关系需要进行深入研究,才能为动脉粥样硬化机制的挖掘提供新思路。
酸性鞘磷脂酶样磷酸二酯酶3B(SMPDL3B)是TLR信号传导的负调节剂。巨噬细胞敲除磷酸二酯酶后导致细胞脂质组成和细胞膜流动性改变,这与TLR配体响应性增加有关[27]。与对照巨噬细胞比较,鞘磷脂合酶(Sgms)1、Sgms 2或丝氨酸棕榈酰转移酶(Sptlc)2缺陷小鼠的巨噬细胞TLR4表达水平降低,TLR4受体转运或再循环障碍[28-29]。尽管脂质组成或脂质筏结构与TLRs间的相互作用关系已确定,但与TLRs结合的脂质物质或蛋白类成分仍不明确。关于TLRs如何在分子水平上与特定的膜脂类物质相互作用尚未见报道。在研究脂质与蛋白质相互作用时,用到的新技术有电子晶体学技术、X-射线晶体学技术,并发现胆固醇分子是G蛋白偶联受体如β2-肾上腺素能受体的整体结构成分,对维持蛋白质稳定性非常重要[30]。除胆固醇外,研究者还发现某些磷脂与跨膜蛋白的特异性相互作用可用于调节TLRs生物功能,例如,TLR3和TLR5的TMD可与鞘磷脂SM C18相结合,通过被膜小泡COPI途径调节蛋白质的分选。
3 结论和展望
在动脉粥样硬化的病理过程中,巨噬细胞具有多重功能,如参与炎症、脂质代谢和细胞凋亡等过程。固有免疫相关的信号通路如TLRs活化对于巨噬细胞泡沫化具有决定性作用,并进一步影响动脉粥样硬化斑块的发展以及并发症的发生。TLR4可通过内源性危险信号如ox-LDL、胆固醇等被激活,诱导巨噬细胞分泌过量的细胞因子和趋化因子,诱发炎症反应和脂质代谢紊乱等。TLR4在动脉粥样硬化中发挥重要作用已被证实,但TLR4活化与巨噬细胞炎症反应中细胞因子分泌以及脂质代谢中胆固醇蓄积的直接作用机制仍不清楚。TLR4作为一种膜识别受体,其生物活性和功能与内源性代谢产物、细胞膜脂质组成、微环境应激的改变密切相关。TLR4作为潜在的治疗和诊断靶点或可改善心血管疾病的治疗。在后续的研究中,我们需要引入新的技术手段,联合免疫学、生物学、生理学与病理学多学科交叉,共同探究TLR4在动脉粥样硬化巨噬细胞泡沫化中的病理机制。
[1] Lim S,Park S.Role of vascular smooth muscle cell in the inflammation of atherosclerosis[J].BMB Rep,2014,47(1):1-7.DOI:10.5483/bmbrep.2014.47.1.285.
[2] Tarkin JM,Joshi FR,Rudd JH.PET imaging of inflammation in atherosclerosis[J].Nat Rev Cardiol,2014,11(8):443-457.DOI:10.1038/nrcardio.2014.80.
[3] Nahrendorf M,Swirski FK.Immunology.Neutrophil-macrophage communication in inflammation and atherosclerosis[J].Science,2015,349(6245):237-238.DOI:10.1126/science.aac7801.
[4] Moore KJ,Tabas I.Macrophages in the pathogenesis of atherosclerosis[J].Cell,2011,145(3):341-355.DOI:10.1016/j.cell.2011.04.005.
[5] Gargiulo S,Gamba P,Testa G,et al.Relation between TLR4/NF-κB signaling pathway activation by 27-hydroxycholesterol and 4-hydroxynonenal,and atherosclerotic plaque instability[J].Aging Cell,2015,14(4):569-581.DOI:10.1111/acel.12322.
[6] Libby P.Inflammation in atherosclerosis[J].Nature,2002,420(6917):868-874.DOI:10.1038/nature01323.
[7] Shibata N,Glass CK.Regulation of macrophage function in inflammation and atherosclerosis[J].J Lipid Res,2009,50(Suppl):S277-S281.DOI:10.1194/jlr.R800063-JLR200.
[8] 耿红莲.TLR4介导氧化型低密度脂蛋白致动脉粥样硬化作用的研究[D].上海:第二军医大学,2006.
[9] 张颖.TLR4与LOX-1信号途径对THP-1细胞炎症因子表达影响及CLI-095抑制动脉粥样硬化研究[D].大连:大连医科大学,2014.
[10] Miller YI,Choi SH,Wiesner P,et al.Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity[J].Circ Res,2011,108(2):235-248.DOI:10.1161/circresaha.110.223875.
[11] Zhong J,Rao X,Oghumu S,et al.Increased expression of dipeptidyl peptidase-4 in atherosclerosis:a role for TLR4/MyD88 signaling[J].Arterioscl Throm Vas Biol,2014,34:A480.
[12] Sindhu S,Al-Roub A,Koshy M,et al.Palmitate-induced MMP-9 expression in the human monocytic cells is mediated through the TLR4-MyD88 dependent mechanism[J].Cell Physiol Biochem,2016,39(3):889-900.DOI:10.1159/000447798.
[13] Kberlin MS,Heinz LX,Superti-Furga G.Functional crosstalk between membrane lipids and TLR biology[J].Curr Opin Cell Biol,2016,39:28-36.DOI:10.1016/j.ceb.2016.01.010.
[14] Levental I,Lingwood D,Grzybek M,et al.Palmitoylation regulates raft affinity for the majority of integral raft proteins[J].Proc Natl Acad Sci USA,2010,107(51):22050-22054.DOI:10.1073/pnas.1016184107.
[15] Kawasaki T,Kawai T.Toll-like receptor signaling pathways[J].Front Immunol,2014,5:461.DOI:10.3389/fimmu.2014.00461.
[16] Fan HC,Fernández-Hernando C,Lai JH.Protein kinase C isoforms in atherosclerosis:pro-or anti-inflammatory[J].Biochem Pharmacol,2014,88(2):139-149.DOI:10.1016/j.bcp.2014.01.006.
[17] Van Eck M,Singaraja RR,Ye D,et al.Macrophage ATP-binding cassette transporter A1 overexpression inhibits atherosclerotic lesion progression in low-density lipoprotein receptor knockout mice[J].Arterioscler Thromb Vasc Biol,2006,26(4):929-934.DOI:10.1161/01.ATV.0000208364.22732.16.
[18] Cavelier C,Lorenzi I,Rohrer L,et al.Lipid efflux by the ATP-binding cassette transporters ABCA1 and ABCG1[J].Biochim Biophys Acta,2006,1761(7):655-666.DOI:10.1016/j.bbalip.2006.04.012.
[19] Yvan-Charvet L,Wang N,Tall AR.Role of HDL,ABCA1,and ABCG1 transporters in cholesterol efflux and immune responses[J].Arterioscler Thromb Vasc Biol,2010,30(2):139-143.DOI:10.1161/ATVBAHA.108.179283.
[20] Tarling EJ,Edwards PA.Dancing with the sterols:critical roles for ABCG1,ABCA1,miRNAs,and nuclear and cell surface receptors in controlling cellular sterol homeostasis[J].Biochim Biophys Acta,2012,1821(3):386-395.DOI:10.1016/j.bbalip.2011.07.011.
[21] Suzuki K,Kawakami Y,Yamauchi K.Impact of TLR 2,TLR 4-activation on the expression of ABCA1 and ABCG1 in raw cells[J].Ann Clin Lab Sci,2017,47(4):436-446.
[22] 欧含笑,郭冰冰,田期先,等.巨噬细胞胆固醇转运相关蛋白研究进展[J].生物化学与生物物理进展,2017,44(2):139-147.
[23] Yvan-Charvet L,Welch C,Pagler TA,et al.Increased inflammatory gene expression in ABC transporter-deficient macrophages:free cholesterol accumulation,increased signaling via toll-like receptors,and neutrophil infiltration of atherosclerotic lesions[J].Circulation,2008,118(18)1837-1847.DOI:10.1161/circulationaha.108.793869.
[24] Zhu X,Owen JS,Wilson MD,et al.Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol[J].J Lipid Res,2010,51(11):3196-3206.DOI:10.1194/jlr.M006486.
[25] Ma L,Dong F,Zaid M,et al.ABCA1 protein enhances Toll-like receptor 4 (TLR4)-stimulated interleukin-10 (IL-10) secretion through protein kinase A (PKA) activation[J].J Biol Chem,2012,287(48):40502-40512.DOI:10.1074/jbc.M112.413245.
[26] Suzuki K,Kawakami Y,Yamauchi K.Impact of TLR 2,TLR 4-activation on the expression of ABCA1 and ABCG1 in raw cells[J].Ann Clin Lab Sci,2017,47(4):436-446.
[27] Heinz LX,Baumann CL,Kberlin MS,et al.The lipid-modifying enzyme SMPDL3B negatively regulates innate immunity[J].Cell Rep,2015,11(12):1919-1928.DOI:10.1016/j.celrep.2015.05.006.
[28] Li Z,Fan Y,Liu J,et al.Impact of sphingomyelin synthase 1 deficiency on sphingolipid metabolism and atherosclerosis in mice[J].Arterioscler Thromb Vasc Biol,2012,32(7):1577-1584.DOI:10.1161/ATVBAHA.112.251538.
[29] Chakraborty M,Lou C,Huan C,et al.Myeloid cell-specific serine palmitoyltransferase subunit 2 haploinsufficiency reduces murine atherosclerosis[J].J Clin Invest,2013,123(4):1784-1797.DOI:10.1172/JCI60415.
[30] Cherezov V,Rosenbaum DM,Hanson MA,et al.High-resolution crystal structure of an engineered human 2-adrenergic G protein-coupled receptor[J].Science,2007,318(5854):1258-1265.DOI:10.1126/science.1150577.
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
湖北农业科学(2022年11期)2022-07-18
科学与财富(2021年33期)2021-05-10
实用肿瘤学杂志(2020年4期)2020-12-08
作文成功之路·小学版(2020年6期)2020-07-27
作文成功之路·小学版(2020年5期)2020-06-11
中成药(2018年9期)2018-10-09
中成药(2018年1期)2018-02-02
中成药(2017年4期)2017-05-17
天津科技大学学报(2016年1期)2016-02-28
