
糖基化终产物对P38MAPK/NF-κB通路诱导的肠道L细胞损伤的影响
2018-06-02 02:06:06周文君张振许宁宁李双喜陈宏
解放军医学杂志 2018年5期
周文君,张振,许宁宁,李双喜,陈宏
胰高血糖素样肽-1(glucagon-like peptide-1,GLP-1)是一种由肠道L细胞分泌的肠促胰素,可作用于胰岛β细胞,促进细胞增殖并抑制其凋亡,促进胰岛素的释放并明显改善胰岛细胞的功能[1-2]。此外,在脂肪细胞中,GLP-1受体激活剂可改善其对胰岛素敏感性,并抑制其增殖[3]。肠促胰岛素效应的减弱可导致胰岛β细胞功能障碍和胰岛素抵抗[4],最终将发展成2型糖尿病,而肠道L细胞的早期炎症损伤可能是GLP-1分泌减少的关键原因[5]。因此,探讨肠道L细胞早期炎症损伤的发生机制对糖尿病的防治研究具有重要意义。晚期糖基化的过程导致糖基化终产物(advanced glycation end products,AGEs)积累,加速了糖尿病、肾衰竭、炎症、神经病变和衰老的进程[6-7]。AGEs作用于糖基化终产物受体(receptor of advanced glycation end products,RAGE)可导致NADPH积累,最终导致核转录因子κB(NF-κB)信号通路的激活,在诱导细胞因子和炎症介质表达的过程中起到关键作用,加剧了糖尿病糖耐量受损阶段的病理改变。而p38MAPK作为NF-κB的重要上游激酶,可被氧化产物激活后诱导NF-κB的活化[8]。2型糖尿病长期的慢性低度炎症可能是肠道L细胞发生早期炎症损伤并导致肠促胰岛素分泌减少的关键原因,与AGEs/RAGE/NADPH/p38MAPK/NF-κB信号通路密切相关。但是,上述通路在肠道L细胞炎症损伤中的作用尚未见报道。本研究旨在探讨AGEs在p38MAPK/NF-κB信号通路诱导的肠道L细胞早期损伤中的作用及机制,以期为研究2型糖尿病的发生发展提供新的思路。
1 材料与方法
1.1 主要试剂及仪器 肠道L细胞株由加拿大多伦多大学Druker实验室的Drucker DJ教授惠赠。10%胎牛血清、DMEM低糖培养基(美国Hyclone公司),兔抗鼠NF-κB p65抗体、鼠抗β-actin单克隆抗体(碧云天公司),兔抗鼠phospho-p38MAPK抗体(美国Santa Cruz公司),HRP标记的羊抗兔IgG抗体(美国R&D公司),HRP标记的羊抗鼠IgG抗体(美国R&D公司),GLP-1、白细胞介素1(IL-1)、白细胞介素6(IL-6)、肿瘤坏死因子α(TNF-α)酶联免疫吸附测定(ELISA)试剂盒(美国Sigma公司),Trizol SYBR及反转录试剂盒(日本TAKaRa公司)。
1.2 方法
1.2.1 细胞培养及分组 将肠道L细胞按文献[9]的方式进行培养,待细胞进入对数生长期后,消化传代。将胎牛血清白蛋白(BSA)和D-葡萄糖溶于PBS中,避光孵育12周制备AGEs-BSA。以荧光分光光度计鉴定AGEs-BSA。采用24孔板铺板细胞,用DMEM低糖培养基预处理,在37℃、5% CO2培养箱中培养6h,使其处于同步化状态,每24h换液。实验分为6组:空白对照组(NC组,即不加入干预因素,仅培养于DMEM低糖培养基中)、BSA对照组(即培养于加入BSA的DMEM低糖培养基中)、AGEs 100μg/ml组(培养于含100μg/ml AGEs的DMEM低糖培养基中)、AGEs 200μg/ml组(培养于含200μg/ml AGEs的DMEM低糖培养基中)、AGEs 300μg/ml组(培养于含300μg/ml AGEs的DMEM低糖培养基中)、AGEs+apocynin组(培养于含200μg/ml AGEs和600μmol/L apocynin的DMEM低糖培养基中[9]),干预方法同前。各组细胞在37℃、5%CO2条件下培养24h后进行各项指标检测。
1.2.2 GLP-1分泌水平检测 采用ELISA法进行检测,其可测范围为4.1~1000pmol/L,批内和批间变异系数分别为5%和12%。按ELISA试剂盒说明书进行操作,在酶标仪上测定波长450nm处的吸光度(OD)值和样本浓度。
1.2.3 p38MAPK、NF-κB p65 mRA水平检测 采用RT-PCR进行检测。使用Trizol试剂从β细胞提取总RNA,再用反转录试剂盒将RNA反转录成cDNA。采用2–ΔΔCt法检测p38MAPK、NF-κB p65的相对基因表达水平。
1.2.4 p-p38MAPK、p38MAPK、NF-κB p65蛋白表达水平检测 采用Western blotting法进行检测。提取细胞总蛋白,用BCA试剂盒测定蛋白浓度。蛋白变性,8% SDS-PAGE电泳,转移至PVDF膜。7.5%脱脂牛奶封闭2h,分别加入特异性一抗phospho-p38MAPK(1:1000)、NF-κB p65(1:1000)和内参β-actin(1:10 000)4℃孵育过夜。洗膜,加入二抗(1:1000羊抗兔IgG抗体)孵育1h,洗膜,采用Bio-Rad凝胶扫描成像系统显影拍照,Quantity One图像软件分析目的蛋白和内参蛋白的灰度值,计算相对灰度值。
1.2.5 炎症因子TNF-α、IL-1、IL-6水平的检测采用ELISA法进行检测。TNF-α的可测范围为50~2450pg/ml,批内和批间变异系数分别为5%和12%;IL-1的可测范围为0.01~1000pg/ml,批内和批间变异系数分别为5%和12%。IL-6的可测范围为0.01~100pg/ml,批内和批间变异系数分别为5%和12%。按ELISA试剂盒说明书进行操作,在酶标仪上测定450nm波长处的OD值和样本浓度。
1.3 统计学处理 采用SPSS 17.0软件进行统计分析。计量资料以±s表示,多组间比较采用单因素方差分析(one-way ANOVA),进一步两两比较采用LSD-t检验。P<0.05为差异有统计学意义。
2 结 果
2.1 各组GLP-1分泌水平的比较 与NC组(92.30±5.10pmol/L)、BSA组(87.75±7.10pmol/L)相比,不同浓度(100、200、300μg/ml)AGEs处理组的GLP-1分泌水平(分别为70.03±4.32、55.28±6.25、38.15±6.10pmol/L)均明显下降(P<0.05),且呈浓度依赖性(P<0.05),采用apocynin抑制NADPH酶的活性后,AGEs+apocynin组GLP-1分泌水平(83.60±7.28pmol/L)较AGEs组明显上升(P<0.05)。
2.2 各组p38MAPK、NF-κB p65 mRNA的表达水平比较 与NC、BSA组相比,随着AGEs浓度的升高,AGEs处理组p38MAPK、NF-κB p65 mRNA有升高的趋势,并与AGEs浓度呈正相关,在200μg/ml和300μg/ml AGEs处理组中的差异有统计学意义(P<0.05)。NADPH酶抑制剂apocynin干预明显抑制了AGEs对p38MAPK、NF-κB p65 mRNA的上调作用,即p38MAPK、NF-κB p65 mRNA水平明显降低(P<0.05,图1)。

图1 AGEs对p38MAPK、NF-κB p65 mRNA水平的调控Fig.1 Effect of AGEs on the level of p38MAPK, NF-κB p65 mRNA
2.3 AGEs对p38MAPK、p-P38MAPK、NF-κB p65蛋白表达的影响 各组p38MAPK蛋白表达水平差异无统计学意义,但p38MAPK蛋白磷酸化(p-p38MAPK)和NF-κB p65蛋白表达差异有统计学意义(Fp-p38MAPK=101.600,FNF-κB=46.666,P=0.000)。其中,各浓度AGEs组p-p38MAPK和NF-κB p65蛋白表达水平较NC组、BSA组明显升高(P<0.05),且呈浓度依赖性,表明随着AGEs浓度的上升,p38MAPK磷酸化水平升高(P<0.05)。而用apocynin抑制NADPH酶的活性后,与AGEs组比较,AGEs+apocynin组p-p38MAPK和NF-κB p65蛋白表达水平明显下降(P<0.05,图2)。
2.4 AGEs对TNF-α、IL-1、IL-6分泌水平的影响
与NC组、BSA组比较,AGEs各浓度组炎症因子TNF-α、IL-1、IL-6分泌水平均上升(P<0.05),呈浓度依赖性,表明AGEs的促炎症作用与其浓度相关。而用apocynin抑制NADPH酶的活性后,TNF-α、IL-1、IL-6表达水平较AGEs组明显下降(P<0.05,表1)。
3 讨 论
AGEs与RAGE结合可引起氧化应激并激活多条细胞信号转导通路,诱发一系列促炎、促凝血反应,促进糖尿病并发症如动脉粥样硬化、肾脏病变的发生与发展[10-11]。本研究前期实验证实,AGEs作用于肠道L细胞,可上调细胞RAGE表达,从而激活NADPH氧化酶,使细胞内ROS生成增多,诱导肠道L细胞的凋亡。此外,AGEs与RAGE结合后不仅导致细胞凋亡,还可诱发炎症反应,现许多研究表明,AGEs可促进炎症因子TNF-α、IL-1、IL-6的分泌,从而损伤细胞,但AGEs的促炎症作用对肠道L细胞的影响尚未见报道[12-13]。GLP-1是肠道L细胞受消化食物刺激后分泌的一种肠促胰素,能够抑制食物的摄取,促进胃排空,改善胰岛素的敏感性,还可作用于α、β细胞,产生双向调节作用。GLP-1受体激动剂以及GLP-1类似物目前已广泛应用于治疗2型糖尿病,成为近年来糖尿病防治领域的重要进展[14]。目前动物研究表明,GLP-1可明显改善胰岛β细胞的功能,促进β细胞增殖并抑制其凋亡[15]。本研究结果显示,随着AGEs浓度的升高,肠道L细胞TNF-α、IL-1、IL-6表达水平升高,即说明了AGEs的促炎症作用。而随着环境中炎症因子水平的上升,又直接影响了肠道L细胞的分泌功能,最终导致细胞GLP-1分泌水平下降。采用apocynin抑制NADPH酶活性后,TNF-α、IL-1、IL-6表达水平明显下降,且GLP-1分泌水平恢复,提示AGEs可诱导肠道L细胞产生炎症损伤,且该作用与NADPH酶的激活有关。
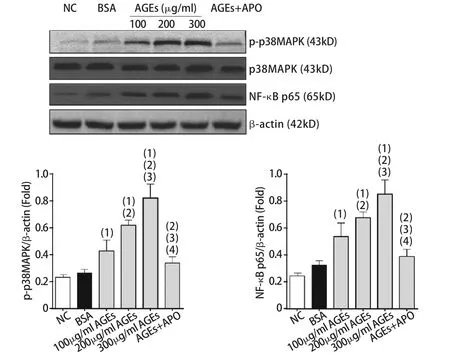
图2 AGEs对p38MAPK、p-p38MAPK、NF-κB p65蛋白表达的影响Fig.2 Effect of AGEs on the protein expression of p38MAPK, p-p38MAPK and NF-κB p65
表1 AGEs对TNF-α、IL-1、IL-6分泌水平的影响(pg/ml,±s)Tab.1 Effect of AGEs on the secretion of TNF-α、IL-1、IL-6 (pg/ml, ±s)

表1 AGEs对TNF-α、IL-1、IL-6分泌水平的影响(pg/ml,±s)Tab.1 Effect of AGEs on the secretion of TNF-α、IL-1、IL-6 (pg/ml, ±s)
APO. Apocynin; (1)P<0.05 compared with NC group and BSA group; (2)P<0.05 compared with 100μg/ml AGEs group; (3)P<0.05 compared with 200μg/ml AGEs group; (4)P<0.05 compared with 300μg/ml AGEs group
Group TNF-α IL-1 IL-6 NC 15.30±1.71 50.67±4.99 43.80±5.94 BSA 16.10±2.15 55.33±6.06 50.40±5.64 100μg/ml AGEs 27.23±4.25(1) 136.50±13.05(1) 95.93±12.29(1)200μg/ml AGEs 38.93±3.62(1)(2) 197.17±12.01(1)(2) 177.93±9.26(1)(2)300μg/ml AGEs 54.47±5.85(1)(2)(3) 287.70±13.75(1)(2)(3) 234.27±19.74(1)(2)(3)AGEs+apocynin 18.40±3.75(2)(3)(4) 68.17±8.72(2)(3)(4) 63.50±7.62(2)(3)(4)
NF-κB作为一种主要的转录因子,可以结合在免疫球蛋白轻链基因增强子上的B细胞特异核蛋白,存在于不同类型细胞中且控制多种基因的表达,这些基因的增强子中均含有NF-κB结合位点,激活的NF-κB能启动这些靶基因的转录,在炎症和自身免疫反应中起重要作用[4,16]。一般情况下,非活化的NF-κB复合物位于细胞质中,该复合物包括功能性亚基p65和抑制性亚基IκBα。功能性亚基可以直接与DNA结合,从而进一步引起基因的转录调控,而抑制性亚基IκBα则可阻止功能性亚基的分离。在应激反应中,IκB激酶(IKK)使抑制型亚基IκBα降解,最终导致功能性亚基p65释放,并向细胞核转移,与DNA结合后引起基因转录调控[17]。p38MAPK是NF-κB的上游激酶,p38MAPK的激活能进一步诱导NF-κB活化,在NF-κB通路的激活中起着重要的作用。Nick等[18]证实LPS刺激中性粒细胞可激活p38MAPK磷酸化,最终调节细胞间黏附及NF-κB活化,并增强TNF-α mRNA及蛋白的表达,提示p38MAPK对NF-κB具有正向调控作用。
有研究表明,血管内皮NADPH氧化酶亚单位Nox-4过度表达,与p22phox一起构成活化的复合物,诱导内皮细胞产生超氧阴离子,并使p38MAPK磷酸化[19]。Sun等[20]研究发现,AGEs可通过RAGE/p38MAPK/NF-κB途径引起人内皮祖细胞功能紊乱,进而促进动脉粥样硬化的形成。Yeh等[21]对人单核细胞的研究表明,AGEs和RAGE结合引起p38MAPK磷酸化,导致NF-κB通路活化,促进炎症因子TNF-α、IL-1、单核细胞趋化蛋白1(MCP-1)的表达和释放,该机制在糖尿病血管并发症的发生、发展中起着重要作用。这提示我们上述AGEs通路的促炎症作用也可能参与肠道L细胞的炎症发生过程,然而目前尚未有研究明确报道。本研究结果提示,随着AGEs处理浓度的提高,p38MAPK磷酸化水平和NF-κB表达量呈剂量依赖性增加,表明NF-κB通路被激活,而用apocynin特异性抑制NADPH氧化酶的活性后,细胞内p38MAPK磷酸化和NF-κB表达量水平下降,说明AGEs与RAGE结合后激活了NADPH氧化酶,继而激活p38MAPK/NF-κB通路,诱导肠道L细胞的炎症损伤,符合之前的研究猜想。
综上所述,本研究发现,AGEs作用于肠道L细胞,与RAGE结合后激活NADPH氧化酶,进而通过p38MAPK/NF-κB途径促进TNF-α、IL-1、IL-6的分泌,使L细胞出现炎症损伤,最终导致GLP-1分泌减少。用apocynin抑制NADPH氧化酶后可减轻AGEs对肠道L细胞的炎症损伤作用,GLP-1分泌可以得到一定程度的恢复。本研究为减轻肠道L细胞的炎症反应以及提高GLP-1分泌水平,从而改善2型糖尿病的发生发展提供了新的思路,为apocynin的应用提供了研究基础,但L细胞的炎症作用于p38MAPK/NF-κB通路的相关机制尚需进一步探索。
[1]Yang L, Yao D, Yang H, et al. Puerarin protects pancreatic β-cells in obese diabetic mice via activation of GLP-1R signaling[J].Mol Endocrinol, 2016, 30(3): 361-371.
[2]Miao XY, Gu ZY, Liu P, et al. The human glucagon-like peptide-1 analogue liraglutide regulates pancreatic beta-cell proliferation and apoptosis via an AMPK/mTOR/P70S6K signaling pathway[J]. Peptides, 2013, 39: 71-79.
[3]Li Y, Du J, Zhu E, et al. Liraglutide suppresses proliferation and induces adipogenic differentiation of 3T3-L1 cells via the Hippo-YAP signaling pathway[J]. Mol Med Rep, 2018, 17(3): 4499-4507.
[4]Haslbeck KM, Schleicher E, Bierhaus A, et al. The AGE/RAGE/NF-(kappa)B pathway may contribute to the pathogenesis of polyneuropathy in impaired glucose tolerance (IGT)[J]. Exp Clin Endocrinol Diabetes, 2005, 113(5): 288-291.
[5]Guglielmi V, Sbraccia P. GLP-1 receptor independent pathways:emerging beneficial effects of GLP-1 breakdown products[J].Eat Weight Disord, 2016, 22(2): 231-240.
[6]Kay AM, Simpson CL, Stewart JA Jr. The role of AGE/RAGE signaling in diabetes-mediated vascular calcification[J]. J Diabetes Res, 2016, 2016: 6809703.
[7]Lu SS, Li XD, Luo SD, et al. Implications of newly-added N-glycosylation mutation of hepatitis B virus S-gene in patients with coexistence of HBsAg and antiHBs[J]. Med J Chin PLA,2016, 41(5): 351-357. [卢姗姗, 李晓东, 罗声栋, 等. HBsAg和抗HBs双阳性患者S基因新增N-糖基化突变的意义[J]. 解放军医学杂志, 2016, 41(5): 351-357.]
[8]Senatus LM, Schmidt AM. The AGE-RAGE Axis: implications for age-associated arterial diseases[J]. Front Genet, 2017, 8: 187.
[9]Hu YH, Zhang Z, Lei L, et al. Advanced glycation end products and its receptor induce apoptosis of L cells through NADPH oxidase mediated signaling pathway[J]. J Pract Med, 2017,33(3): 358-362. [胡颖辉, 张振, 雷蕾, 等. 糖基化终产物通过其受体激活NADPH氧化酶及其下游通路诱导L细胞的凋亡[J]. 实用医学杂志, 2017, 33(3): 358-362.]
[10]Wang XL, Yu T, Yan QC, et al. AGEs promote oxidative stress and induce apoptosis in retinal pigmented epithelium cells RAGE-dependently[J]. J Mol Neurosci, 2015, 56(2): 449-460.
[11]Yamagishi S, Takeuchi M, Inagaki Y, et al. Role of advanced glycation end products (AGEs) and their receptor (RAGE) in the pathogenesis of diabetic microangiopathy[J]. J Int Med Res,2003, 23(4): 129-134.
[12]You J, Zhu ML, Peng W, et al. Advanced glycation endproducts induce the expression of TNF-α through RAGE in Jurkat cells[J]. Chin J Immun, 2012, 28(7): 594-598. [游捷, 朱明理,彭伟, 等. AGEs通过RAGE促进Jurkat细胞分泌肿瘤坏死因子-α[J]. 中国免疫学杂志, 2012, 28(7): 594-598.]
[13]Yanagida M, Jung G, Tanaka Y, et al. Serum proteome analysis in patients with rheumatoid arthritis receiving therapy with etanercept, a chimeric tumor necrosis factor-alpha receptor[J].Int J Rheum Dis, 2012, 15(5): 486-495.
[14]Bastien-Dionne PO, Valenti L, Kon N, et al. Glucagon-like peptide 1 inhibits the sirtuin deacetylase SirT1 to stimulate pancreatic β-cell mass expansion[J]. Diabetes, 2011, 60(12): 3217-3222.
[15]Lee YA, Wang MY, Du XQ, et al. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice[J]. Diabetes,2011, 60(2): 391-397.
[16]Mao X, Su Z, Mookhtiar AK. Long non-coding RNA: a versatile regulator of the nuclear factor -κB signaling circuit[J].Immunology, 2016, 150(4): 379-388.
[17]Gilmore TD. Introduction to NF-kappaB: players, pathways,perspectives[J]. Oncogene, 2006, 25(51): 6680-6684.
[18]Nick JA, Avdi NJ, Young SK, et al. Selective activation and functional significance of p38alpha mitogen-activated protein kinase in lipopolysaccharide-stimulated neutrophils[J]. J Clin Invest, 1999, 103(6): 851-858.
[19]Goettsch C, Goettsch W, G, Seebach J, et al. Nox4 overexpression activates reactive oxygen species and p38 MAPK in human endothelial cells[J]. Biochem Biophys Res Commun, 2009,380(2): 355-360.
[20]Sun C, Liang C, Ren Y, et al. Advanced glycation end products depress function of endothelial progenitor cells via p38 and ERK 1/2 mitogen-activated protein kinase pathways[J]. Basic Res Cardiol, 2009, 104(1): 42-49.
[21]Yeh CH, Sturgis L, Haidacher J, et al. Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factor-κB transcriptional activation and cytokine secretion[J]. Diabetes, 2001, 50(6): 1495-1504.
猜你喜欢
大自然探索(2024年1期)2024-03-19 19:01:03
日用电器(2022年7期)2022-09-07 07:05:00
当代水产(2021年10期)2022-01-12 06:20:42
中国果业信息(2021年7期)2021-12-01 20:20:32
保健医苑(2021年7期)2021-08-13 08:47:54
今日农业(2020年22期)2020-12-14 16:45:58
凤凰生活(2019年8期)2019-08-16 02:00:38
食品安全导刊(2016年7期)2016-05-14 12:39:11
中国病理生理杂志(2015年8期)2015-12-21 12:38:06
医学研究杂志(2015年3期)2015-06-10 06:41:52
