
丝裂原活化蛋白激酶磷酸酶1在糖尿病大鼠肾组织细胞外基质重构中的作用
2018-05-31 07:41王丽晖吴广礼黄旭东杨新军汪晶华陈云爽
解放军医药杂志 2018年5期
王丽晖,吴广礼,林 静,黄旭东,杨新军,汪晶华,陈云爽,赵 维
肾小球硬化是糖尿病肾病(DN)的基本病理改变,细胞外基质(ECM)重构是导致肾小球硬化的主要原因之一[1]。高糖状态下蛋白质非酶糖化增高,肾脏血流动力学紊乱、细胞增殖和凋亡及细胞通路的激活等参与了DN肾小球硬化的发生和发展[2]。研究表明,丝裂原激活的蛋白激酶(MAPK)信号转导通路的激活,尤其是p38 MAPK信号转导蛋白活化后,可以激活多种核转录因子,影响目的基因表达,在一定程度上加速了DN的病变过程[3]。有关p38 MAPK信号通路在DN发病机制中的研究较多,但其在DN肾小球硬化中的表达如何调整,受何种因素调节,目前尚未有进一步的研究。丝裂原活化蛋白激酶磷酸-1(MKP-1)可通过使MAPK脱磷酸化进而使MAPK灭活,是MAPK信号通路的特异性负性调节因子[4]。在DN的ECM重构中MKP-1的作用,和其与p38 MAPK相互作用,目前尚不得知。本研究拟通过糖尿病大鼠肾组织中MKP-1蛋白及mRNA的表达情况,观察MKP-1与p38 MAK信号转导途径之间的相互作用,以揭示DN时ECM重构可能发生的机制,为进一步防治DN提供理论依据。
1 材料与方法
1.1实验动物 SPF级雄性Wistar大鼠60只,河北省实验动物中心提供,许可证号:1607934。1~12周龄,体重120~150 g,性周期4~5 d。饲养条件:按性周期一致性原则,每3只合并一笼,室温21~22℃,湿度67%~75%,光照比例1:1。
1.2实验仪器和试剂 链脲佐菌素(STZ,sigma公司,美国);兔抗大鼠MKP-1和鼠抗大鼠p-p38 MAPK 购自美国Santa Cruz 公司;兔抗大鼠纤维粘连蛋白(Laminin,LN)和小鼠抗大鼠纤维粘连蛋白(Fibronectin,FN)均购自美国Neomarker 公司;免疫组化试剂盒(北京中山生物技术有限责任公司);考马斯亮蓝试剂盒购自南京建成生物工程研究所;聚偏二氟乙烯膜( PVDF)购自美国merckmillipor公司; RT-PCR试剂为美国Promega 公司;Trizol购自美国Sigma公司;其他试剂均为分析纯。
1.3实验方法
1.3.1糖尿病肾病大鼠模型的构建:按65 mg/kg腹腔单剂量注射链脲佐菌素(STZ,溶于0.1 mol/L枸橼酸缓冲液, PH=4.5)建立糖尿病模型,48 h后大鼠尾尖取血测定血糖,取尿测尿糖。血糖≥16.7 mmol/L,尿糖3+~4+者确定为建模成功。建模成功的大鼠随机分为1、2、4、8和12周组,每组10只,实验观察期间大鼠自由进食饮水,不使用降糖药物和胰岛素。另选取10只大鼠为正常对照组,只注射相同体积的枸橼酸缓冲液(pH=4.5、0.1 mol/L)。
1.3.2肾组织标本收集:糖尿病模型组制模成功后1、2、4、8、12周分别处死动物,切取肾脏,去掉被膜,滤纸吸干血迹后称重,留取部分肾组织迅速置于-70℃液氮中提取RNA和蛋白质,留作反转录聚合链连反应(RT-PCR)和Western实验检测;部分置于新鲜配制的4%多聚甲醛溶液(0.01 mol/L PBS 配制)进行固定,留作常规肾脏病理和免疫组化检测。
1.3.3免疫组化染色检测肾皮质MKP-1、p-p38 MAPK和LN的表达:用SP法(链霉素抗生素蛋白-过氧化物酶联结法),切片厚4 μm,常规脱蜡水化,一抗为免抗鼠MKP-1和LN多克隆抗体(1∶50稀释),鼠抗p-p38 MAPK单克隆抗体(1∶100稀释),二抗为生物素化羊抗兔或鼠IgG(1∶100稀释),以PBS代替一抗作为阴性对照,DAB显色,光镜观察阳性信号,阳性部位呈棕黄色。免疫组化结果应用HPIAS-1000高清晰度彩色病理图文分析系统进行分析,LN阳性结果胞质着色,分别测量出每个肾小球LN的着色阳性面积、积分光密度(IOD)和肾小球面积,以前两个参数与肾小球面积比表示该成分的相对含量和表达强度。p-p38 MAPK、MKP-1等胞核着色的免疫组化结果检测每个肾小球视野内阳性胞核细胞数与所有肾小球细胞数之比,作为阳性百分比。每组分析6个标本,每个标本切片分析20个完整的肾小球,20个肾小球均值代表一只大鼠某种成分在肾小球中的相对含量和表达强度。20个肾小球“阳性百分比”代表一只大鼠某种成分在肾小球中的表达,以IOD表示其表达强度。
1.3.4Western blotting检测肾皮质细胞MKP-1和p38 MAPK的活性:取-70℃保存的肾皮质约100 mg,加入预冷的蛋白裂解液0.5 ml。取100 μg蛋白加入上样缓冲液,应用12%SDS-聚丙烯酰胺凝胶进行电泳。电泳后转PVDF膜,分别与兔抗大鼠MKP-1和p-p38MAPK多克隆抗体(1∶100),4℃ 过夜后,再度洗膜后加入1∶2500稀释的辣根过氧化物酶标记的羊抗兔IgG二抗,37℃,2 h。洗膜后加增强化学发光(ECL, Santa Cruz公司) 试剂,将PVDF膜放入X光片暗盒,压片,显影,定影。用美国Kodak公司1D数码成像分析系统软件对Western条带进行相对定量分析。
1.3.5RT-PCR检测肾皮质MKP-1和FN mRNA的表达:采用Trizol提取肾皮质RNA,应用紫外可见分光光仪测定RNA的纯度和含量。取总RNA 2 μg作为模板,在反转录酶(US)催化下合成cDNA,以适量cDNA 为模板在Taq DNA聚合酶催化下在聚合酶链反应(PCR)扩增仪进行PCR扩增。MKP-1,FN及GAPDH引物均由北京奥科生物工程有限公司合成,使用的基因引物如下:MKP-1 F:5'- ACA GGG CAG AAG GGA AAG GAC -3', R:5' - GCT GAA AGC CTG CTC TGG GTC -3' 产物:263bp;FN:F:5′CTGAACCAGCCTACGGATGACT3′R:5′CCGTCGTCATAACACGTTGCT3′产物305b;GAPDH: F:5′TATCGGACGCCTGGTTAC3′R:5′CTGTGCCGTTGAACTTGC3′ 产物140bp。PCR反应条件:94℃,5 min;95℃,1 min;采用退火温度分别为55.9℃、54.9℃和56.9℃,1 min;72℃,1 min;扩增32个循环,最后72℃,10 min。取5ul PCR产物在2%琼脂糖凝胶进行电泳,观察条带情况。应用凝胶图像分析系统(UVP GD S800,U S) Labworks 4.5软件测定IOD,并以GAPDH基因表达量为内参照,以目的产物与GAPDH的积分光密度比值表示mRNA相对含量。

2 结果
2.1免疫组化检测结果 MKP-1主要在肾小球的内皮细胞、系膜细胞及肾小管上皮细胞的胞核中表达,在对照组肾小球和肾小管的细胞核着色较强,在糖尿病组表达明显减少(见图1)。p38 MAPK主要表达在肾小球内皮细胞、系膜区及脏层上皮细胞的胞质内,偶有近端和远端肾小管上皮细胞表达,与对照组比较糖尿病组的表达较强(见图2)。LN的阳性染色主要分布于毛细血管基膜、肾小球囊壁和肾小管基膜和系膜区,在肾间质中也有表达,与对照组比较,糖尿病组表达增强(见图3)。图像分析结果表明,与对照组比较,糖尿病组MKP-1表达在1~8周表达量明显减少(P<0.01);第12周表达量与对照组无统计学差异(P>0.05)。糖尿病组p38 MAPK在 1~8周时表达增强(P<0.01),在 12周时与对照组比较无统计学差异(P<0.01)。LN在糖尿病组的表达从4~12周均明显增高(P<0.01)。见表1。
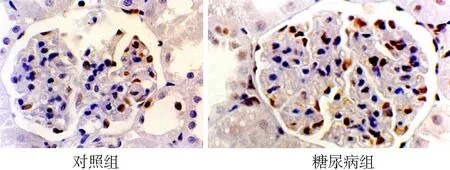
图1免疫组化检测MKP-1在两组大鼠肾组织中的表达(DAB×400)
图2免疫组化检测p-p38MAPK在两组大鼠肾组织中的表达(DAB×200)

图3免疫组化检测Lamain在两组大鼠肾组织中的表达(DAB×400)

表1 两组大鼠肾组织中p-p38 MAPK 、MKP-1和LN的表达
注:p-p38 MAPK为磷酸化p38丝裂原活化蛋白激酶,MKP-1为丝裂原活化蛋白激酶磷酸酶-1,LN为层粘连蛋白;与对照组相比,bP<0.01
2.2Western blotting检测结果 糖尿病组1~8周MKP-1蛋白的表达量低于对照组(P<0.01),但在糖尿病12周时与对照组比较差异无统计学意义(P>0.05)。p38蛋白激酶的活性从糖尿病1周时开始升高,在4、8周时达到高峰(P<0.01);在12周时与对照组差异无统计学意义(P≥0.05)。见图 4,表2。
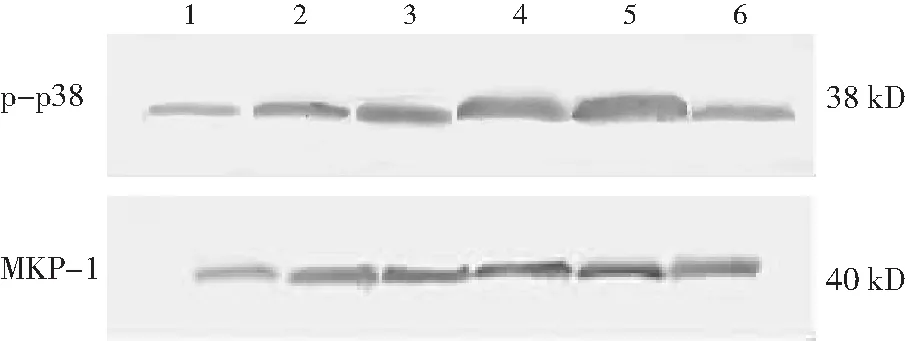
图4两组大鼠肾皮质P-p38MAPK和MKP-1的动态变化
1代表对照组 ,2、3、4、5、6代表糖尿病组 1、2、4、8、12周
表2 两组大鼠肾皮质中MKP-1和p-p38 MAPK的表达
注:MKP-1为丝裂原活化蛋白激酶磷酸酶,p-p38 MAPK为磷酸化p38丝裂原活化蛋白激酶;与对照组相比,bP<0.01
2.3RT-PCR检测结果 MKP-1 mRNA的表达在糖尿病1-8周时低于对照组(P<0.01),12周时与对照组无统计学差异(P>0.05)。与对照组比较,FN mRNA在糖尿病第2周开始升高,随着时间的延长其表达越来越强(P<0.01)。而各磷酸甘油醛脱氢酶(GAPDH)的表达几乎保持恒定(表3,图5)。
表3 两组大鼠肾皮质中MKP-1和FN mRNA的表达
注:MKP-1为丝裂原活化蛋白激酶磷酸酶,FN为纤维粘连蛋白;与对照组相比,bP<0.01
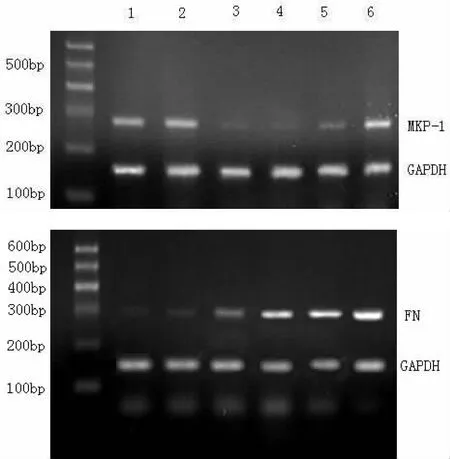
图5两组大鼠肾皮质中MKP-1和FNmRNA的动态变化
3 讨论
糖尿病肾病时ECM重构是糖尿病肾小球硬化的基础;既往研究表明,高糖有可能是导致ECM重构的直接因素;血流动力学异常、氧化应激、细胞因子等均可导致ECM重构的发生[5]。MAPK家族在各种刺激因子的作用下,通过使核转录因子磷酸化,参与细胞增殖、分化、转化及凋亡的调节;p38 MAPK被激活后进入细胞核,使核转录调节因子活化,从而调节相应目的基因转录,在一定程度上导致和加速了DN的发生、发展[6]。认为p38 MAPK是细胞信息传递的交汇点或共同通路[7]。本研究结果显示,糖尿病组p38 MAPK的磷酸化水平在DM 1周时开始升高,在4~8周时达到高峰,在DM 12周时其活性与对照组无明显差异。而作为ECM主要成分的LN在糖尿病组从4~12周表达明显增高,并随病程的延长而表达增强。提示p38蛋白激酶活化后,可以使LN的表达明显增强,p38MAPK通路在糖尿病大鼠的ECM的重构中发挥一定作用。我们既往研究发现[8],体外培养的大鼠系膜细胞在高糖状态下p38 MAPK信号转导蛋白磷酸化水平提高,可以上调结缔组织生长因子(CTGF)mRNA的表达,从而刺激细胞上清液中FN和LM的分泌。Rane等[9]观察了体外培养的肾小管上皮细胞,在高糖状态下凋亡基因增强,凋亡细胞增多,提示p38 MAPK信号转导通路不仅在肾小球系膜细胞中发挥作用,同时参与肾小管上皮细胞的凋亡过程。目前针对DN的治疗主要是控制血压、控制血糖、改善肾脏微循环等,但也难以逆转DN的进程。所以选择抑制p38 MAPK信号转导通路的激活,有可能在糖尿病肾病的预防和治疗中发挥一定作用。
MKP-1是MAPKs的天然负性调节因子,对MAPKs 的去磷酸化作用有重要的意义[10]。MAPKs 调节位点中保守的苏氨酸或酪氨酸基团可被双特异性蛋白激酶磷酸化激活,而双特异性蛋白磷酸酶可使同样位点的苏氨酸或酪氨酸基团去磷酸化,从而使活化的MAPKs主要被蛋白磷酸酶去磷酸化而失活[11]。Kato等[12]观察心房利钠肽(ANP)对体外培养的肾小球足细胞的影响时发现,ANP可增加MKP-1的活性,同时阻断p38 MAPK的活性,导致凋亡基因Bax和 Bax/Bcl2的表达下降。p38 MAPK的阻断剂FR167653,可以降低血压,降低尿蛋白排出,减轻节段硬化,足细胞受损和细胞凋亡。因此,ANP通过阻断p38 MAPK通路,增加MKP-1的活性起到肾脏保护功能。蛋白磷酸酶的活性是蛋白激酶活性的100~1000 倍,故将注意力放在蛋白磷酸酶的生理作用上。一般认为MKP-1 的特异性选择底物是p38,在一些情况下也可能是JNK 或ERK[13]。
本实验结果显示,糖尿病模型大鼠在注射链脲佐菌素1周后,MKP-1活性及mRNA 表达较对照组下降,并持续到第8周,在第12周时其蛋白及mRNA的表达下降。而p38 MAPK的活性在1周时开始升高,FN、LN水平在第4周升高,p38 MAPK的活性及FN、LN的表达升高持续到12周。MKP-1的活性下降早于FN和LN。提示糖尿病状态下,大鼠肾组织MKP-1的蛋白和mRNA的表达均下降,而且MKP-1活性的下降晚于p38 MAPK活性的上升,提示高糖可以使MKP-1的合成减少,降解增加。文献报道,高糖培养大鼠的肾小球系膜细胞MKP- 1 蛋白的表达明显下降,而p38 MAPK 活性增加[14]。提示糖尿病肾病时p38 MAPK 活化是由于磷酸化增强、MKP- 1 的脱磷酸化减弱所致。
一般认为, MKP-1是一种细胞核酶,能快速调整MAPK的去磷酸化,短时间内发挥作用。Fang等[4]观察提示越早期应用胰岛素可使MKP-1表达增加,降低p38 MAPK活性,有效减轻肾脏肥大的发生。Gorostizaga等[15]曾发现当近端肾小管上皮细胞暴露于高浓度白蛋白培养液下, MKP-1水平在15 min即快速升高,在6 h时达到顶峰,随即下降。尿白蛋白通过基因转录的激活可使MKP-1 mRNA水平短暂升高(1 h为对照组的3倍)。尿中白蛋白不仅触发MAPK的活化也紧紧地上调MKP-1表达。所以推测MKP-1 的短半衰期与MAPK对其磷酸化作用[16]。Winiarska等[17]研究结果提示细胞活动中MKPs 的表达是继发于MAPK 激活后,即MKPs 的表达是细胞对MAPK 通路激活的一种反馈。MKP-1在低渗应激下可激活p38 MAPK, 诱导β和γ-肾小管上皮细胞钠通道蛋白的表达。PD98059作为MEK抑制剂,可以诱导MKP-1的表达从而抑制p38MAPK的活化。本实验结果也表明,模型大鼠在第2周时p38 MAPK的活性表达增强,MKP-1的蛋白表达下降,提示MKP-1对p38 MAPK的负调节是继发于p38 MAPK激活后,同时这种磷酸化作用可以抑制蛋白酶对MKP-1 的降解,从而增强其稳定性,MKP-1 表达也受到MAPKs 的负反馈调节。MKP-1 与MAPKs 之间相互作用,相互影响。
综上所述, MKP-1 是p38 MAPK 的负性调节因子,可以对p38 MAPK 的主要通路进行调节,并抑制其下游反应。激活后可使糖尿病大鼠肾组织的细胞外基质的含量降低,在DN的ECM重构中发挥保护作用,可减轻DN病理改变,延缓DN发生和发展。如果能发现能使提高MKP-1 的表达的药物,可对抗高糖对细胞外基质重构的影响,从而为临床治疗DN提供理论基础。
[]
[1] Soni H, Adebiyi A. Urotensin II-induced store-operated Ca2+entry contributes to glomerular mesangial cell proliferation and extracellular matrix protein production under high glucose conditions[J].Sci Rep, 2017,22,7(1):18049.
[2] Wang S, Zhao X, Yang S,etal. Salidroside alleviates high glucose-induced oxidative stress and extracellular matrix accumulation in rat glomerular mesangial cells by the TXNIP-NLRP3 inflammasome pathway[J].Chem Biol Interact, 2017,278(1):48-53.
[3] Wang R M, Wang Z B, Wang Y,etal. Swiprosin-1 Promotes Mitochondria-Dependent Apoptosis of Glomerular Podocytes via p38 MAPK Pathway in Early-Stage Diabetic Nephropathy[J]Cell Physiol Biochem, 2018,45(3):899-916.
[4] Fang D,Guan H,Liu J,etal. Early intensive insulin therapy attenuates the p38 pathway in the renal cortex and indices of nephropathy in diabeticrats[J].Endocr J, 2012,59(1):81-90.
[5] Li J, Bao L, Zha D,etal. Oridonin protects against the inflammatory response in diabetic nephropathy by inhibiting the TLR4 /p38 -MAPK and TLR4/NF-κB signaling pathways[J].Int Immunopharmacol, 2017,55(1):9-19.
[6] 王丽晖,段惠军,史永红,等.p38 丝裂原活化蛋白激酶在糖尿病大鼠肾组织细胞外基质重构中的作用[J].解放军医学杂志,2005,30(4):310-313.
[7] Yeda X,Shaoqing L,Yayi H,etal. Dexmedetomidine protects against renal ischemia and reperfusion injury by inhibiting the p38-MAPK/TXNIP signaling activation in streptozotocin induced diabeticrats[J].Acta Cir Bras,2017,32(6):429-439.
[8] 王丽晖,段惠军,吴广礼,等.高糖诱导大鼠系膜细胞CTGF mRNA的表达及机制探讨[J].中国病理生理杂志,2009,25(8):1644-1646.
[9] Rane M J, Song Y, Jin S,etal. Interplay between Akt and p38 MAPK pathways in the regulation of renal tubular cell apoptosis associated with diabetic nephropathy[J].Am J Physiol Renal Physiol, 2010,298(1):F49-61.
[10] Thiel G, Rössler O G. Resveratrol stimulates AP-1-regulated gene transcription[J].Mol Nutr Food Res, 2014,58(7):1402-1413.
[11] Tsai S F, Hsieh C C, Wu M J,etal. Novel findings of secreted cyclophilin A in diabetic nephropathy and its association with renal protection of dipeptidyl peptidase 4 inhibitor[J].Clin Chim Acta, 2016,463(1):181-192.
[12] Kato Y,Mori K, Kasahara M,etal. Natriuretic peptide receptor guanylyl cyclase-A pathway counteracts glomerular injury evoked by aldosterone through p38 mitogen-activated protein kinase inhibition[J].Sci Rep, 2017,7:44642.
[13] Akhtar S, Al-Zaid B, El-Hashim A Z,etal. Impact of PAMAM delivery systems on signal transduction pathways in vivo: Modulation of ERK1/2 and p38 MAP kinase signaling in the normal and diabetic kidney[J].Int J Pharm, 2016,514(2):353-363.
[14] Jung Y J, Lee A S, Nguyen-Thanh T,etal. SIRT2 Regulates LPS-Induced Renal Tubular CXCL2 and CCL2 Expression[J].J Am Soc Nephrol, 2015,26(7):1549-1560.
[15] Gorostizaga A, Mori Sequeiros García M M,etal. Modulation of albumin-induced endoplasmic reticulum stress in renal proximal tubule cells by upregulation of mapk phosphatase-1[J].Chem Biol Interact, 2013,206(1):47-54.
[16] Zhou X, Liu R, Duan S,etal. High Glucose Enhances oxLDL-Induced Apoptosis in Human Renal Proximal Tubular Epithelial Cells Largely via Inducing Lectin-Like ox-LDL Receptor-1[J].Pharmacology, 2016,98(1-2):20-28.
[17] Winiarska K, Jarzyna R, Dzik J M,etal. ERK1/2 pathway is involved in renal gluconeogenesis inhibition under conditions of lowered NADPH oxidase activity[J].Free Radic Biol Med, 2015,81:13-21.
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
世界科学技术-中医药现代化(2021年8期)2021-12-21
天津医科大学学报(2021年4期)2021-08-21
科学(2020年2期)2020-08-24
天津医科大学学报(2019年6期)2019-08-13
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
医学研究杂志(2015年9期)2015-07-01
医学研究杂志(2015年3期)2015-06-10
中国医学科学院学报(2013年6期)2013-03-11
