
铁镍物质的量比对双金属催化剂性能的影响
2018-05-30 06:18安晓倩张古承张静卫陈默周鹏徐浩叶倩周冠宇杨富花
中南大学学报(自然科学版) 2018年5期
安晓倩,张古承,张静,卫陈默,周鹏,徐浩,叶倩,周冠宇,杨富花
铁镍物质的量比对双金属催化剂性能的影响
安晓倩,张古承,张静,卫陈默,周鹏,徐浩,叶倩,周冠宇,杨富花
(四川大学建筑与环境学院,四川 成都,610065)
采用溶胶−凝胶法制备铁镍双金属氧化物((Fe-NiO)催化剂以避免均相体系金属离子污染的问题。制备不同物质的量比的铁镍双金属催化剂,利用X线衍射(XRD)、X线光电子能谱(XPS)、扫描电子显微镜(SEM)、傅里叶变换红外线光谱(FT−IR)对催化剂表面特性进行表征;同时以邻苯二甲酸二甲酯(DMP)的降解率为指标衡量其异相催化臭氧(O3)的能力,并且重复使用5次以探究催化剂的稳定性。研究结果表明:制备的上述5种催化剂均表现出优越的催化性能,实验进行5 min后DMP降解率分别为85.93%,63.33%,83.92%,78.58%和80.93%;投加催化剂后,O3分解率与DMP降解率具有相似的趋势。Fe-NiO催化剂表面组成主要为Fe3O4,NiFe2O4和NiO;Fe-NiO催化剂微粒主要由0.1~0.3 μm颗粒集聚形成,并产生较多孔洞;Fe-NiO催化剂表面存在表面羟基(—OH),由此推测催化过程主要为多价态金属元素及—OH共同作用的结果。
高级氧化;异相催化;双金属氧化物;臭氧
当今自然水体中的有机污染物越来越复杂,通过传统的水处理技术难以将其高效降解。同时,某些残留的有毒物质会对人体的健康产生潜在的威胁。高级氧化技术(AOPs)因能达到高效、快速降解有机物污染物的目的而得以快速发展。近年来,催化臭氧工艺因其对难降解有机物具有高效处理能力以及对水质产生较低的负面影响而得到广泛关注[1−3]。臭氧催化过程中产生的羟基自由基(∙OH)是一种强氧化剂,对大多数有机污染物具有较强的降解能力[4−5],能够克服单独臭氧工艺对有机污染物的选择性问题。这种技术不需要辅助加热或光照系统,可直接应用于实际水处理工程[6]。ANIPSITAKIS等[7−11]研究发现过渡金属具有较强的催化臭氧的能力,相对于均相催化,过渡金属元素氧化物异相催化在保留其催化效能的基础上避免了金属离子污染的问题[12]。MURUGANANDHAM等[13]合成的氧化锌对2−乙氧基乙酸乙酯具有良好的催化降解效果。DONG等[9]将-MnO2作为新型水处理催化剂催化臭氧降解有机污染物,取得良好效果。此外,SONG等[14]探讨了不同制备条件对二氧化钛降解苯酚活性的影响。铁、镍、铜、钴也是较为常用的制备催化剂的过渡金属元素[15−17]。与单一金属元素氧化物相比,对双金属氧化物的研究相对较少。根据已有文献报道,在适宜条件下制备的双金属氧化物与单一金属氧化物相比具有更好的催化性能。LI等[18]发现Pd-Ce金属氧化物异相催化臭氧降解丙酮酸比单一氧化物表现出更加优良的催化活性。同时,REN等[19]发现双金属氧化物对过硫酸氢盐也具有显著的催化效果。可见,双金属氧化物在水处理领域中具有广阔的应用前景。不同的制备条件对催化剂表面组成以及表面官能团的影响不同[20−22]。本文作者探讨制备过程中金属元素物质的量比对铁镍双金属氧化物(Fe-NiO)催化剂的影响,以邻苯二甲酸二甲酯(DMP)为目标物,衡量不同制备条件下催化剂的催化效能。通过X线衍射(XRD)和X线光电子能谱(XPS)表征Fe-NiO催化剂的组成成分以及化学状态,并且利用傅里叶变换红外线光谱(FT−IR)探讨催化剂表面存在的官能团,采用扫描电子显微镜 (SEM)探讨其表面形貌特征。
1 设备材料与方法
1.1 实验材料
DMP 购自西格玛−奥德里奇(美国);六水硝酸镍(Ni(NO3)2∙6H2O)、九水硝酸铁(Fe(NO3)3∙9H2O)、叔丁醇(TBA)、二水磷酸二氢钠(NaH2PO4∙2H2O)、十二水磷酸氢二钠(Na2HPO4∙12H2O)和亚硫酸钠(Na2SO3)均购自成都市科龙化工试剂厂(中国成都),以上所有的药品均为分析纯,实验所用溶液由超纯水配制;新鲜鸡蛋在当地市场购得(中国成都);反应体系pH由磷酸缓冲液调节。
1.2 催化剂制备
Fe-NiO催化剂利用鸡蛋清通过溶胶−凝胶法(sol–gel process)制备[19, 23]。将铁镍混合金属离子溶液缓慢加入鸡蛋清中,剧烈搅拌至均匀混合。将混合液置于80℃水浴中加热形成凝胶,静置陈化后烘干,在500℃下焙烧得到Fe-NiO催化剂。改变铁镍混合液中金属离子物质的量比,分别为(Fe3+):(Ni2+)= 1:4,1:2,1:1,2:1,4:1(其中,为物质的量,mol),得到不同制备条件下的催化剂。
1.3 实验过程与分析
催化O3氧化DMP的实验在平底烧瓶内进行。反应体系水量为500 mL,反应体系温度为(18±1)℃,磷酸缓冲液调节pH=7.1。O3以干燥O2(纯度≥99%)为气源,通过臭氧发生器(HTU−500SE型,AZCO公司,加拿大)生成,O3的浓度通过紫外可见分光光度计(UV−1800型,美普达仪器有限公司,中国上海) 在波长254 nm处测量[24]。在溶液中加入Fe-NiO催化剂和 DMP溶液瞬间开始计时,取样时样品经玻璃纤维滤膜 (孔径为0.7 μm,Whatman,英国) 过滤后,立即转移至含有过量Na2SO3溶液的试管中终止反应。
DMP的浓度由高效液相色谱仪 (HPLC,Waters e2695,Waters公司,美国) 分析测定,色谱柱型号为Waters C18(5 μm),检测器波长为230 nm,流动相为甲醇和纯水混合液(甲醇与水体积比为1:1),流量为1.0 mL/min。O3分解实验体系中不投加DMP溶液,保持其余控制条件一致进行实验,O3的浓度通过靛蓝法利用紫外可见分光光度计进行检测[25]。
2 结果与讨论
2.1 XPS表征
图1所示为Fe-NiO催化剂的XPS谱。图1(a)中Fe 2p轨道的结合能710.9 eV和724.8 eV分别对应Fe3O4[26]及NiFe2O4中的Fe的结合能。Fe3O4存在形式为FeO∙Fe2O3,即Fe存在+2与+3价态。
图1(b)中Ni 2p轨道上+3价对应电子结合能为861.4 eV,而855.4 eV与873.4 eV均对应+2价的Ni的电子结合能[27],可见Fe-NiO催化剂表面Fe和Ni化合价均存在+2和+3这2种价态。氧化还原反应的本质为电子的转移,催化剂中多价态金属元素的存在会加速转移过程,从而产生强氧化性自由基[1, 28−29]。
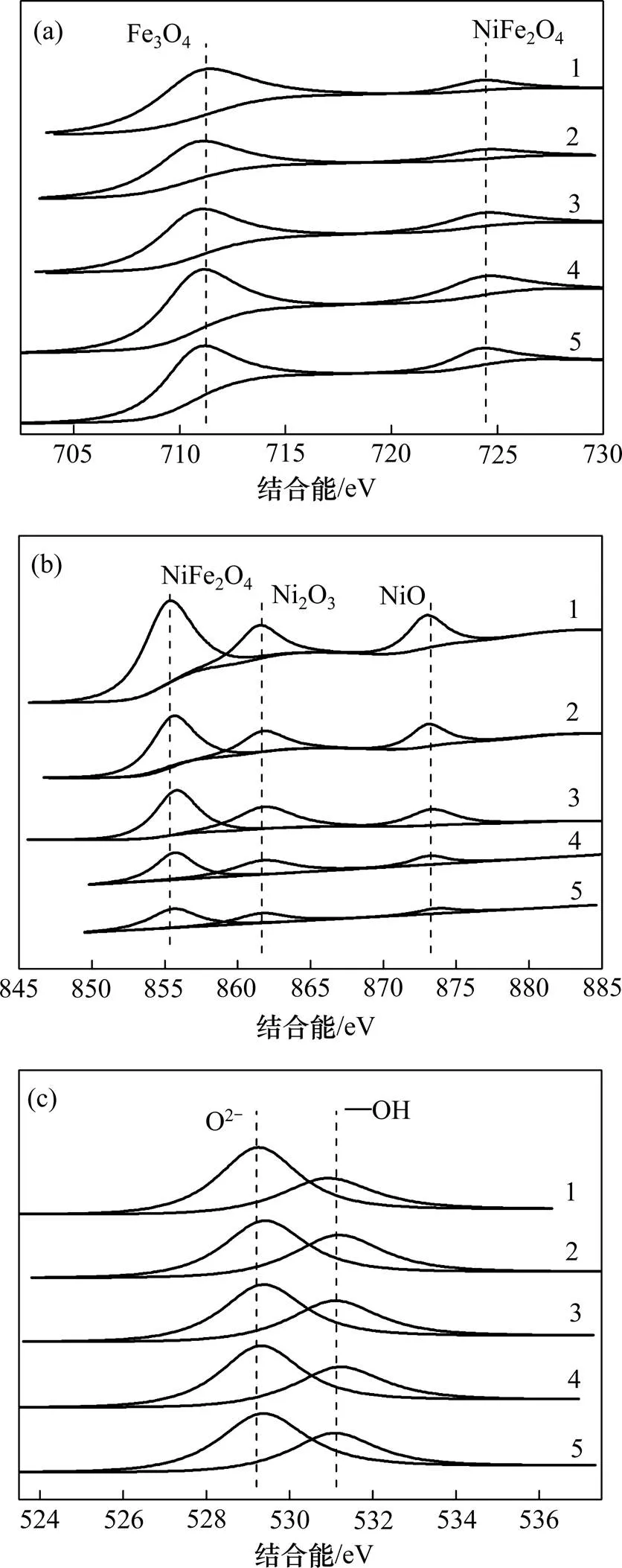
(a) Fe分谱;(b) Ni分谱;(c) O分谱
图1(c)中O 1s 轨道出现2个峰,其中529.4~ 530.0 eV处的峰对应金属氧化物中的晶格氧XPS峰,531.25~531.8 eV处的峰则对应催化剂表面的—OH中的氧XPS峰。研究表明表面—OH在催化过程中具有重要作用,是影响催化效率的主要因素之一[30]。表面—OH的存在使金属离子处于配位不饱和的状态,进而作为活性位点吸附水溶液中有机污染物,并且与溶解的O3作用引发其链式分解,最终导致∙OH的生成[31−32]。
总体而言,很难直接通过观察比较出不同催化剂中峰强度的细微变化,因此,由峰面积计算Fe-NiO催化剂中NiFe2O4,Fe3O4和−OH摩尔分数,结果如表1所示。

表1 不同Fe-NiOx催化剂中NiFe2O4, Fe3O4和—OH摩尔分数
2.2 XRD表征
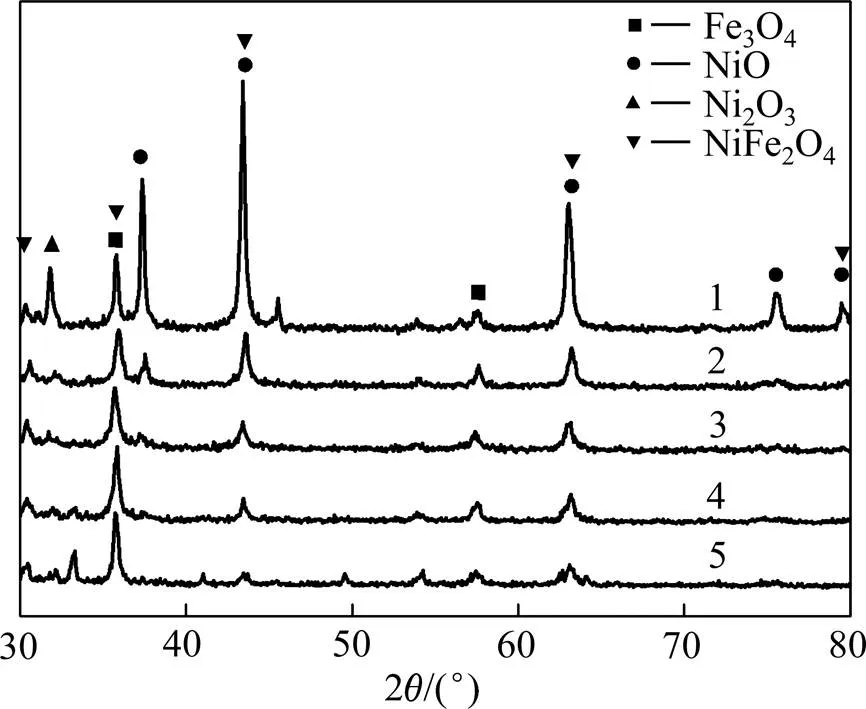
图2所示为不同催化剂的XRD谱。从图2可看出:催化剂表面金属氧化物主要为Fe3O4,NiFe2O4,Ni2O3和NiO,与XPS表征结果吻合。由图1(b)发现,当(Fe3+):(Ni2+)由4:1变化至1:4时,Ni的峰强度增强,其中Ni2+增强尤为明显,表明Fe-NiO催化剂中Ni2+逐渐增多;当(Fe3+):(Ni2+)=1:4时,NiO和NiFe2O4的衍射峰变得更尖锐,表明催化剂结晶程度较好。可见催化剂中Ni2+增加,相应生成的产物NiO和NiFe2O4具有更好的锐化度,衍射峰更强烈。
2.3 FT−IR表征
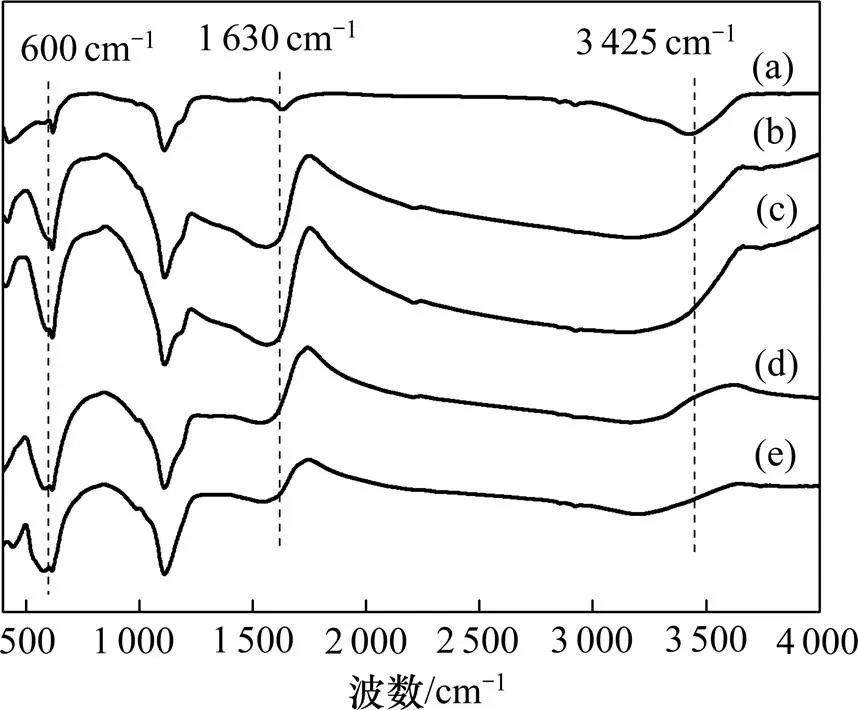
不同条件制备的催化剂的FT−IR表征结果如图3所示。由图3可以看出:催化剂表面具有2个表面羟基(—OH)的吸附峰,首先,在3 425 cm−1处的峰对 应为M—OH (M为Fe或Ni)结构中氢键拉伸作用[28],其次,1 628~1 640 cm−1处出现的峰归因于催化剂表面吸附水的弯曲振动作用。结合表1中—OH摩尔分数分析,制备过程中金属元素物质的量比的改变并未导致催化剂表面—OH摩尔分数发生显著变化。此外,600 cm−1附近出现的吸附峰则对应混合金属氧化物NiFe2O4中四面体位置固有的拉伸振动峰[33],再次表明催化剂中NiFe2O4的存在与XRD和XPS所示结果一致。
n(Fe3+):n(Ni2+):1—1:4;2—1:2;3—1:1;4—2:1;5—4:1。
2.4 SEM表征
图4所示为不同Fe-NiO催化剂的SEM图,图4中5种催化剂均由细小颗粒物组成。由图4可见:颗粒直径为0.1~0.3 μm,这些细小颗粒互相集聚从而形成较大的微粒。相较于其他图像,图4(b)所示的细小颗粒分布极为均匀,集聚现象更明显,使Fe-NiO催化剂微粒间空隙较小;图4(a)所示细小颗粒则形成较为稳固的微观结构,颗粒集聚形成的微粒互相支撑形成较多的孔洞。
n(Fe3+):n(Ni2+):(a) 1:4;(b) 1:2;(c) 1:1;(d) 2:1;(e) 4:1

n(Fe3+):n(Ni2+):(a) 1:4;(b) 1:2;(c) 1:1;(d) 2:1;(e) 4:1
2.5 Fe-NiOx催化剂的催化性能
加入不同制备条件下所得Fe-NiO催化剂,比较体系中DMP降解情况以分析催化剂的活性,如图5所示。由图5可知:当反应体系中不投加催化剂时,反应进行5 min后DMP降解率为38.52%;而投加催化剂后DMP降解率得到显著提升,Fe-NiO催化剂可显著提升反应体系的氧化能力,促进目标物DMP降解。在体系中投加(Fe3+):(Ni2+)=1:4的催化剂,DMP降解率达85.93%;相比之下,投加(Fe3+):(Ni2+)=1:2的催化剂,DMP降解率略低,为63.33%,显然前者的催化性能更优越。
已有研究发现,—OH可能是O3分解为自由基的活性位点,催化剂的催化性能可能与其表面的—OH摩尔分数呈正相关[11, 34]。结合图1(c)和图3可知:本研究制备的不同的Fe-NiO催化剂表面均含有—OH。—OH的存在可为O3分解为自由基提供反应场所,从而使DMP降解率较单独的O3体系得到提升。此外,金属氧化物的多价态转换促进界面电子转移也可能会促进有机物的快速降解[1, 28]。
比较表1 中(Fe3+):(Ni2+)=1:2,(Fe3+):(Ni2+)= 1:4的数据可知,后者—OH相对较少而含有较多NiFe2O4和Fe3O4,但 DMP降解率却有所提升,表明—OH摩尔分数并不是影响异相催化高级氧化体系氧化能力的主要因素。图5中(Fe3+):(Ni2+)=1:4与(Fe3+):(Ni2+)=2:1时DMP降解率相差不多,后者含有更多的NiFe2O4和Fe3O4以及较少的—OH,可见虽然金属元素多价态间转换会促进高级氧化体系中自由基的生成[19],但也不是决定性因素。异相催化高级氧化过程是一个多种因素共同作用的复杂过程,需要进行更深入研究。

图5 不同Fe-NiOx催化剂的催化性能
2.6 Fe-NiOx催化剂使用次数对DMP降解的影响
图6所示为不同Fe-NiO催化剂第2~5次使用、反应5 min后DMP的降解情况。由图6可知:不同Fe-NiO均具有较稳定的催化性能,5种Fe-NiO催化剂在第2次使用时DMP降解率与图5相比基本保持不变,分别为61.38%,78.48%,81.32%,78.46%和82.47%。第4次、第5次使用的催化效果出现较明显下降,但与图5中不投加催化剂相比仍具有一定的催化活性,表明制备出的催化剂稳定性较强,多次使用仍具有较好的催化效果。
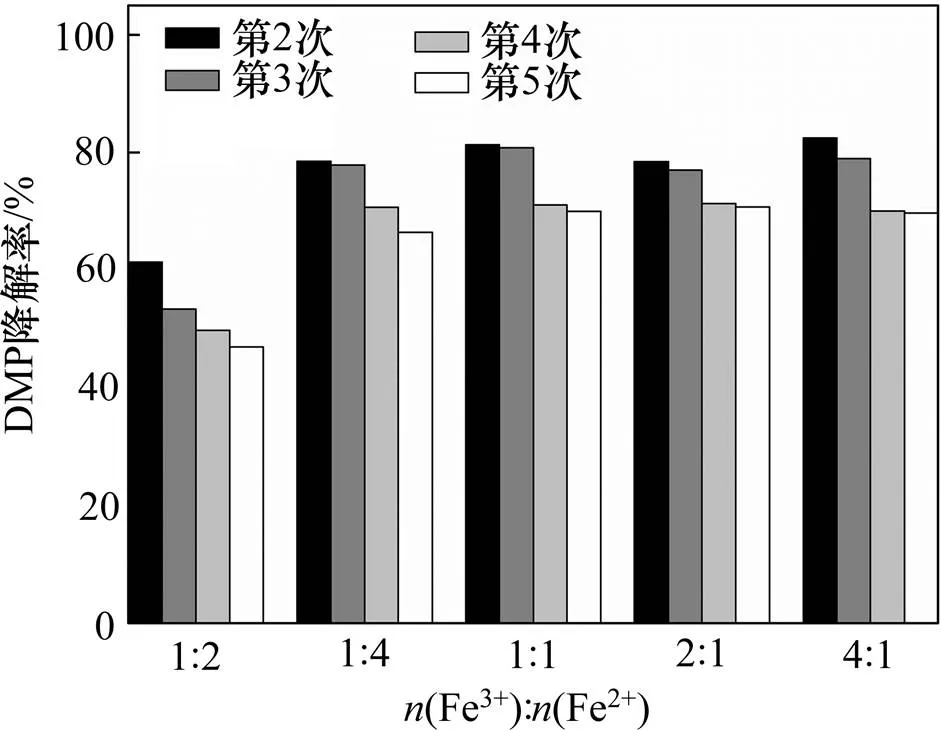
图6不同使用次数Fe-NiOx催化剂的催化性能
2.7 Fe-NiOx催化剂对O3分解的影响
图7所示为投加不同Fe-NiO催化剂时O3的分解率。从图7可以看出:当不投加催化剂时,10 min 后O3分解率仅为22.22%,而投加不同Fe-NiO催化剂后,O3分解率均得到有效提升,并且其变化趋势与图5显示DMP降解率相似,表明体系中DMP的降解主要是因为投加催化剂引起O3分解。
2.8 催化过程中活性物种类
根据文献报道,叔丁醇(TBA)与O3的反应速率常数为0.03 L/(mol∙s),而与∙OH的反应速率常数为2.2×108L/(mol∙s)[35−37],其可作为∙OH的抑制剂,以证明其为O3/Fe-NiO体系中引起DMP降解的氧化物种。选用制备条件为(Fe3+):(Ni2+)=1:4的催化剂进行验证实验,结果如图8所示。由图8可知:与只投加O3时相比,投加催化剂后,DMP降解率由38.52%提升至85.93%,投加TBA后分别下降至21.68%与24.05%。而只投加Fe-NiO催化剂DMP降解率为3.13%,催化剂的吸附降解可以忽略,可见体系中DMP降解主要是由O3分解产生∙OH引起的。

n(Fe3+):n(Ni2+):1—1:4;2—1:2;3—1:1;4—2:1;5—4:1;6—无催化剂。
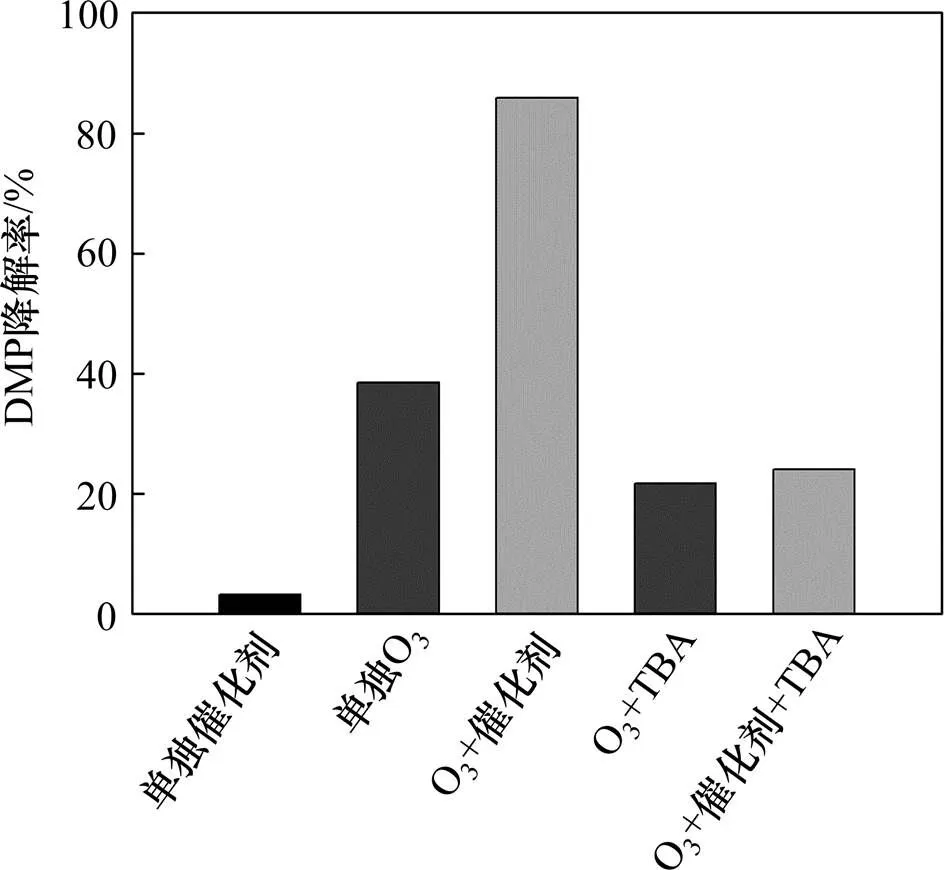
图8 投加TBA后DMP分解率
3 结论
1) Fe-NiO催化剂对O3体系具有显著的催化效果,投加Fe-NiO催化剂后,体系的氧化能力得到显著提升,并且DMP降解率与O3分解率具有相似的变化趋势。
2) 不同Fe与Ni物质的量比制得的催化剂表面组成主要为NiFe2O4,Fe3O4,Ni2O3和NiO,但催化剂结晶度以及成分摩尔分数发生变化。
3) SEM结果表明采用溶胶−凝胶法制备出的Fe-NiO催化剂为纳米级,主要由0.1~0.3 μm颗粒集聚而产生较多孔洞,从而形成催化剂的主体结构。
4) XPS与FT−IR结果验证了Fe-NiO催化剂表面存在—OH,不同Fe与Ni物质的量比会影响最终得到的催化剂中有效成分—OH,NiFe2O4和Fe3O4。
5) Fe-NiO催化剂中表面—OH摩尔分数并非影响异相催化能力的唯一因素,金属元素的多价态间的转换也会促进O3分解产生∙OH,从而提升DMP降解率。
[1] XING Shengtao, HU Chun, QU Jiuhui, et al. Characterization and reactivity of MnOsupported on mesoporous zirconia for herbicide 2,4-D mineralization with ozone[J]. Environmental Science & Technology, 2008, 42(9): 3363−3368.
[2] NIE Yulun, HU Chun, LI Nengneng, et al. Inhibition of bromate formation by surface reduction in catalytic ozonation of organic pollutants over β-FeOOH/Al2O3[J]. Applied Catalysis B: Environmental, 2014, 147(1): 287−292.
[3] TRAPIDO M, HIRVONEN A, VERESSININA Y, et al. Ozonation, ozone/UV and UV/H2O2degradation of chlorophenols[J]. Ozone Science & Engineering, 1997, 19(1): 75−96.
[4] ANDREOZZI R, INSOLA A, CAPRIO V, et al. The use of manganese dioxide as a heterogeneous catalyst for oxalic acid ozonation in aqueous solution[J]. Applied Catalysis A: General, 1996, 138(1): 75−81.
[5] LEGUBE B, LEITNER N. Catalytic ozonation: a promising advanced oxidation technology for water treatment[J]. Catalysis Today, 1999, 53(1): 61−72.
[6] FU H X, LEITNER N, LEGUBE B. Catalytic ozonation of chlorinated carboxylic acids with Ru/CeO2−TiO2catalyst in the aqueous system[J]. New Journal of Chemistry, 2002, 26(11): 1662−1666.
[7] ANIPSITAKIS G P, DIONYSIOU D D. Radical generation by the interaction of transition metals with common oxidants[J]. Environmental Science & Technology, 2004, 38(13): 3705−3712.
[8] HUANG Ruihuan, YAN Huihua, LI Laisheng, et al. Catalytic activity of Fe/SBA−15 for ozonation of dimethyl phthalate in aqueous solution[J]. Applied Catalysis B: Environmental, 2011, 106(1/2): 264−271.
[9] DONG Yuming, YANG Hongxiao, HE Kun, et al. β-MnO2nanowires: a novel ozonation catalyst for water treatment[J]. Applied Catalysis B: Environmental, 2009, 85(3/4): 155−161.
[10] PULLABHOTLA V S R R, RAHMAN A, JONNALAGADDA S B. Selective catalytic Knoevenagel condensation by Ni-SiO2supported heterogeneous catalysts: an environmentally benign approach[J]. Catalysis Communications, 2009, 10(4): 365−369.
[11] YANG Yixin, MA Jun, QIN Qingdong, et al. Degradation of nitrobenzene by nano-TiO2catalyzed ozonation[J]. Journal of Molecular Catalysis A: Chemical, 2007, 267(1/2): 41−48.
[12] SANCHEZ−POLO M, RIVERA−UTRILLA J, VON GUNTEN U. Metal-doped carbon aerogels as catalysts during ozonation processes in aqueous solutions[J]. Water Research, 2006, 40(18): 3375−3384.
[13] MURUGANANDHAM M, WU J J. Synthesis, characterization and catalytic activity of easily recyclable zinc oxide nanobundles[J]. Applied Catalysis B: Environmental, 2008, 80(1/2): 32−41.
[14] SONG Shuang, LIU Zhiwu, HE Zhiqiao, et al. Impacts of morphology and crystallite phases of titanium oxide on the catalytic ozonation of phenol[J]. Environmental Science & Technology, 2010, 44(10): 3913−3918.
[15] PINES D S, RECKHOW D A. Solid phase catalytic ozonation process for the destruction of a model pollutant[J]. Ozone Science & Engineering, 2003, 25(1): 25−39.
[16] ZHAO Lei, SUN Zhizhong, MA Jun.Novel relationship between hydroxyl radical initiation and surface group of ceramic honeycomb supported metals for the catalytic ozonation of nitrobenzene in aqueous solution[J]. Environmental Science & Technology, 2009, 43(11): 4157−4163.
[17] YUAN Lei, SHEN Jimin, CHEN Zhonglin, et al. Role of Fe/pumice composition and structure in promoting ozonation reactions[J]. Applied Catalysis B: Environmental, 2016, 180: 707−714.
[18] LI Weiwei, QIANG Zhimin, ZHANG Tao, et al. Kinetics and mechanism of pyruvic acid degradation by ozone in the presence of PdO/CeO2[J]. Applied Catalysis B: Environmental, 2012, 113/114: 290−295.
[19] REN Yueming, DONG Qing, FENG Jing, et al. Magnetic porous ferrospinel NiFe2O4: a novel ozonation catalyst with strong catalytic property for degradation of di-n-butyl phthalate and convenient separation from water[J]. Journal of Colloid and Interface Science, 2012, 382(1): 90−96.
[20] DE DIEGO L F, GAYÁN P, GARCÍA−LABIANO F, et al. Impregnated CuO/Al2O3oxygen carriers for chemical-looping combustion: avoiding fluidized bed agglomeration[J]. Energy & Fuels, 2005, 19(5): 1850−1856.
[21] CHAN S C, BARTEAU M A. Preparation of highly uniform Ag/TiO2and Au/TiO2supported nanoparticle catalysts by photodeposition[J]. Langmuir, 2005, 21(12): 5588−5595.
[22] MAZILLE F, SCHOETTL T, PULGARIN C. Synergistic effect of TiO2and iron oxide supported on fluorocarbon films(part 1): effect of preparation parameters on photocatalytic degradation of organic pollutant at neutral pH[J]. Applied Catalysis B: Environmental, 2009, 89(3/4): 635−644.
[23] MAENSIRI S, MASINGBOON C, BOONCHOM B, et al. A simple route to synthesize nickel ferrite (NiFe2O4) nanoparticles using egg white[J]. Scripta Materialia, 2007, 56(9): 797−800.
[24] VALDES H, MURILLO F, MANOLI J, et al. Heterogeneous catalytic ozonation of benzothiazole aqueous solution promoted by volcanic sand[J]. Journal of Hazardous Materials, 2008, 153(3): 1036−1042.
[25] BADER H, HOIGNE J. Determination of ozone in water by the indigo method[J]. Water Research, 1981, 15(4): 449−456.
[26] ORLÉANS R B C. Database for surface spectroscopies as XPS, AES and UPS[EB/OL]. [2016−10−23]. www.lasurface. com/database/elementxps.php.
[27] WANG Z, CHOU H, WU J C S, et al. CO2photoreduction using NiO/InTaO4in optical-fiber reactor for renewable energy[J]. Applied Catalysis A: General, 2010, 380(1/2): 172−177.
[28] LÜ Aihua, HU Chun, NIE Yulun, et al. Catalytic ozonation of toxic pollutants over magnetic cobalt and manganese co-doped γ-Fe2O3[J]. Applied Catalysis B: Environmental, 2010, 100(1/2): 62−67.
[29] SUI Minghao, LIU Jia, SHENG Li. Mesoporous material supported manganese oxides (MnO/MCM−41) catalytic ozonation of nitrobenzene in water[J]. Applied Catalysis B: Environmental, 2011, 106(1/2): 195−203.
[30] MACHOCKI A, IOANNIDES T, STASINSKA B, et al. Manganese-lanthanum oxides modified with silver for the catalytic combustion of methane[J]. Journal of Catalysis, 2004, 227(2): 282−296.
[31] JOSEPH Y, RANKE W, WEISS W. Water on FeO(111) and Fe3O4(111): adsorption behavior on different surface terminations[J]. Journal of Physical Chemistry B, 2000, 104(14): 3224−3236.
[32] ZHANG Tao, MA Jun. Catalytic ozonation of trace nitrobenzene in water with synthetic goethite[J]. Journal of Molecular Catalysis A: Chemical, 2008, 279(1): 82−89.
[33] FLOREA M, ALIFANTI M, PARVULESCU V I, et al. Total oxidation of toluene on ferrite-type catalysts[J]. Catalysis Today, 2009, 141(3/4): 361−366.
[34] ZHANG Tao, LI Chunjuan, MA Jun, et al. Surface hydroxyl groups of synthetic alpha-FeOOH in promoting (OH)−O−center dot generation from aqueous ozone: property and activity relationship[J]. Applied Catalysis B: Environmental, 2008, 82(1/2): 131−137.
[35] ANDREOZZI R, CAPRIO V, INSOLA A, et al. Advanced oxidation processes (AOP) for water purification and recovery[J]. Catalysis Today, 1999, 53(1): 51−59.
[36] HOIGNE J, BADER H. Role of hydroxyl radical reactions in ozonation processes in aqueous solutions[J]. Water Research, 1976, 10(5): 377−386.
[37] HOIGNE J, BADER H. Role of hydroxyl radical reactions of ozone with organic and inorganic compounds in water 2: dissociating organic compounds[J]. Water Research, 1983, 17(2): 185−194.
(编辑 刘锦伟)
Effect of Fe-Ni bimetal oxides with different element molar ratios on catalytic activity
AN Xiaoqian, ZHANG Gucheng, ZHANG Jing, WEI Chenmo, ZHOU Peng, XU Hao, YE Qian, ZHOU Guanyu, YANG Fuhua
(School of Architecture & Environment, Sichuan University, Chengdu 610065, China)
Bimetal Fe-Ni oxides (Fe-NiO) catalysts prepared by sol−gel method were synthesized to avoid the pollution caused by metal ions in homogeneous system. Different bimetal catalysts were prepared with different mole ratios of Fe and Ni, and the surface properties were characterized by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS), scanning electron microscope (SEM) and Fourier transform infrared spectroscopy (FT−IR). The degradation rate of dimethyl phthalate (DMP) was measured as the target that revealed the activity of catalysts to activated O3, and the stability of bimetal catalysts was investigated after being used for 5 times. The results show that the prepared 5 catalysts work well and the degradation rates of DMP are 85.93%, 63.33%, 83.92%, 78.58% and 80.93% in 5 min, respectively. The O3decomposition rates have the similar trend, indicating that the degradation rate of DMP is almost caused by the decomposition of O3. Fe3O4, NiFe2O4and NiO are the main phases in the surface of Fe-NiOcatalysts. The average grain size of Fe-NiOcatalysts is in the range of 0.1−0.3 μm and many holes are generated with these grain assembled. There are hydroxyl groups (—OH) on the surface of catalysts. Therefore, it can be inferred that the catalytic process is possible due to the combined action of multi-valence of metal elements and —OH.
advanced oxidation process; heterogeneous catalysis; bimetal oxides; ozone
10.11817/j.issn.1672-7207.2018.05.011
X131.2
A
1672−7207(2018)05−1103−07
2017−05−28;
2017−07−18
国家自然科学基金资助项目(51508353);成都市科技惠民项目(2015-HM01-00279-SF) (Project(51508353) supported by the National Natural Science Foundation of China; Project(2015-HM01-00279-SF) supported by the Science and Technology Huimin Project of Chengdu)
张静,博士,副教授,从事水深度处理研究;E-mail: zjing428@163.com
猜你喜欢
化工管理(2022年13期)2022-12-02
中国钱币(2022年1期)2022-08-23
中国金属通报(2022年6期)2022-06-22
环境保护与循环经济(2021年7期)2021-11-02
陶瓷学报(2021年4期)2021-10-14
陶瓷学报(2021年1期)2021-04-13
陶瓷学报(2020年6期)2021-01-26
海洋通报(2020年5期)2021-01-14
航天工业管理(2020年9期)2020-12-28
试题与研究·高考理综化学(2016年3期)2017-03-28
