
视神经脊髓炎谱系疾病合并自身免疫性疾病诊治体会
2018-05-21 05:39李艾帆杨改清姜晓蕊袁树华
中风与神经疾病杂志 2018年3期
李艾帆, 杨改清, 姜晓蕊, 燕 燕, 袁树华
视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disease,NMOSD)是中枢神经系统脱髓鞘疾病,多与结缔组织病、系统性红斑狼疮等自身免疫性疾病共存[1],临床症状的不典型常给诊断带来困难,若能及时诊治,效果较为理想。现将我科近期诊治的视神经脊髓炎谱系疾病合并自身免疫性疾病2例,结合相关文献总结报道如下。
1 临床资料
病例1,女,54岁,农民,以“胸背部感觉异常20 d,四肢无力7 d”为主诉于2016年11月4日入院。入院前20 d患者无诱因出现右上肢发痒、疼痛,症状逐渐波及前胸后背,皮肤表面无皮疹、红肿及破溃,在当地按皮肤病治疗(具体不详)无效果,1 w前病情继续发展,出现下腹部及双下肢麻木、感觉异常,并出现右下肢无力,可站立,行走困难,症状渐加重,发展至四肢无力,上肢可抬举,持物不牢,下肢不能站立行走,右侧较重,无晨轻暮重,无发热,无头晕、头痛,无视物模糊、视物成双,无四肢抽搐,无言语不清及意识障碍,无大小便障碍,门诊以“四肢无力查因”收入院。病来神志清,精神差,睡眠、进食可,大小便正常。既往体健,无高血压、糖尿病及感染病史。家族史:1子患有系统性红斑狼疮。入院查体:体温36.5 ℃,脉搏93次/min,呼吸19次/min,血压145/88 mmHg,心肺听诊无异常,双下肢无水肿。神经系统查体:神清,语利,颅神经检查正常,右下肢肌力2级,右上肢肌力4+级,左上肢肌力5-级,左下肢4+级,肌张力正常,双侧腱反射活跃(),双侧肢体病理征阳性,左侧霍夫曼征阳性,右侧指鼻试验欠稳准,T2以下浅感觉减退,深感觉正常,脑膜刺激征阴性。辅助检查:颈胸髓MRI:延髓、C2-T5平面脊髓异常信号(见图1);颈椎退行性改变并C4/5、C5/6、C6/7椎间盘突出。头部MRI示延髓异常信号,考虑非肿瘤性病变,炎性脱髓鞘病变可能性大。心电图示下壁心肌呈缺血型改变。胸部CT:左肺上叶陈旧病灶。肌电图示左上肢尺神经及双下肢H反射传导异常;双侧听觉传导通路未见明显异常;双侧视神经传导通路异常;右下肢深感觉传导通路异常。脑脊液:外观清亮,压力160 mmH2O,pandy试验(+),蛋白 39 mg/dL,AQP4结果阳性,寡克隆区带阴性,QAIb升高(提示血脑屏障破坏)。血常规示白细胞 10.48×109/L,中性细胞比率 83.30 %,血脂、肝肾功能、血糖、甲功三项、传染病筛查、凝血功能、肿瘤筛查均正常。抗核抗体谱:抗SS-A抗体(阳性)、抗Ro-52抗体(阳性)、抗SS-B抗体(阳性)、核糖体p蛋白(阳性)。该患者没有感染、肿瘤及代谢性疾病,临床诊断:视神经脊髓炎谱系疾病(NMOSD)合并系统性红斑狼疮。给予丙种球蛋白0.4 g/kg·d,连用5 d,甲泼尼龙冲击治疗12 d后,改为强的松80 mg口服,每2 w减5 mg逐渐减量,免疫抑制剂环磷酰胺0.4 g,治疗1 m出院时患者四肢无力好转,能下床活动,感觉障碍平面下降,6 m后复诊,患者四肢肌力进一步好转,生活可以自理,感觉障碍仍存在,较前好转,复查颈胸MRI较前明显好转(见图2)。
病例2,女,38岁,以“四肢麻木1 w余,加重2 d余”为主诉,于2017年2月3日收入院。入院前1 w患者无诱因出现四肢麻木不适,呈进行性加重,无视物不清,无发热,咳嗽、腹泻,无肢体无力,无复视,无恶心、呕吐,无头晕、头痛,无言语不清及意识障碍,无四肢抽搐,入院前2 d四肢麻木加重,伴右侧肢体无力,持续不能缓解,遂至我院门诊以“(1)脊髓病变;(2)吉兰-巴雷?”收入我科。病来神志清,精神差,进食、睡眠可,大、小便正常,体重无明显变化,既往体健,无高血压病、糖尿病、冠心病病史,无药物过敏史。入院查体:体温36.0 ℃,脉搏72次/min,呼吸18次/min,血压130/90 mmHg,心肺听诊无异常,双下肢无水肿。神经系统查体:意识清,言语流利,颅神经检查正常,四肢肌力5级,肌张力正常,双侧腱反射(),双侧病理征阴性,闭目难立征阴性,深浅感觉减退,脑膜刺激征阴性。辅助检查:颈胸MRI示:C1-T1椎体水平异常信号(见图3),考虑脊髓病变;颈4/5、5/6、6/7椎间盘突出,颈椎病。抗核抗体谱:抗SS-A抗体(+),抗Ro-52抗体(+),抗SS-B抗体(+),抗核小体抗体(+)。心电图示:窦性心律,下壁心肌呈缺血型改变;胸部DR示:心肺
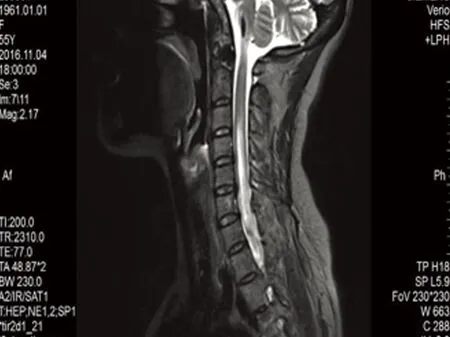
图1 治疗前T2序列

图2 治疗后6 mT2序列

图3 治疗前T2序列

图4 治疗后1 m T2序列
膈未见明显异常。叶酸 4.89 ng/ml减低,红细胞 3.62×1012/L,血红蛋白 110.00 g/L,电解质、肝 肾功能、血糖、尿常规、传染病筛查、甲功三项、血栓六项及心肌酶谱均正常。腰穿脑脊液压力145 mmH2O,蛋白 52 mg/dL,pandy试验 阳性(+),细胞数正常。AQP4、寡克隆区带检查均为阳性,补体C3 0.527 g/L,补体C4 0.099 g/L,免疫球蛋白A 4.9 g/L,免疫球蛋白G 33.0 g/L,补体C1q 250 mg/L,根据患者的临床症状、脊髓MRI改变、免疫学检查,支持诊断为视神经脊髓谱系疾病合并系统性红斑狼疮。治疗:应用激素冲击12 d后,改为醋酸泼尼松片 80 mg 口服,每14 d减量5 mg,治疗1 m,四肢麻木症状基本消失,复查颈MRI:脊髓内病变减轻(见图4),激素缓慢减量,病情稳定出院。
2 讨 论
视神经脊髓炎(neuromyelitis optica,NMO)是指双侧视神经炎和脊髓炎在短期内相继发生的中枢神经系统脱髓鞘疾病,最早于1984年由Devic提出。近年来有些学者认为,MNO应独立于经典的MS,但对于MNO是独特的脱髓鞘疾病还是MS的一个亚型一直存在争议。随着影像学和神经免疫学的发展,较多的学者认为两者在临床、影像、实验室指标等方面有诸多不同。2007年Wingerchuk[2]提出了视神经谱系疾病(neuromyelitis optica spectrum disease,NMOSD)的概念,特指一组在发病机制与视神经脊髓炎相近,又与视神经脊髓炎不同的一种疾病,其临床症状不典型,识别相对困难。2015年国际NMOSD诊断标准[3],在NMOSD诊断中提出血清或脑脊液AQP-4-Ab阳性为主要的诊断依据,并将NMOSD分为AQP4抗体阳性和AQP4抗体阴性二大类。
AQP4-IgG阳性NMOSD诊断标准[4]:(1)必须具备一项核心临床特征;(2)免疫学指标:AQP4抗体阳性;(3)排除其他诊断。AQP4-IgG阴性或未检测的NMOSD诊断标准[4]:(1)临床诊断条件:①至少有两项核心临床特征,其中视神经炎为必备条件,同时具备急性脊髓炎或延髓最后区综合征;②空间多发;③符合MRI附加条件。(2)AQP4抗体阴性或无条件检测或未检测;(3)排除其他诊断。核心临床特征主要包括:视神经炎(伴有或不伴视力下降、视觉诱发电位异常);急性长节段脊髓炎(不完全性横断性脊髓炎);最后区综合征,发作性或顽固性呃逆、恶心呕吐;其他脑干综合征;睡眠障碍;间脑综合征等。
AQP4抗体阴性或未检测的NMOSD的MRI附加条件:脑MRI正常或仅有非特异性白质病变;(1)视神经MRI长T2信号或增强的T1信号大于1/2视神经长度,或病变累及视交叉。(2)急性脊髓炎,脊髓病变大于三个连续椎体节段,如果患者有脊髓炎病史,其MRI检查发现相应脊髓萎缩大于3个连续椎体节段。(3)最后区综合征:延髓背侧/最后区病变。(4)急性脑干综合征:脑干室管膜周围病变。
NMO-IgG是视神经和脊髓的中枢神经系统炎性脱髓鞘疾病的特异性抗体,NMO-IgG的靶抗原是AQP-4-Ab。AQP-4-Ab对NMO诊断的敏感性为73%,特异性为91%,已作为NMO特异性抗体纳入NMO的诊断标准中[5]。有研究显示,AQP-4-Ab阳性仅见于视神经脊髓炎谱系疾病(NMOSD)患者以及SLE/SS合并NMOSD患者中,表明两者均为自身免疫性疾病,可以合并存在,而AQP-4抗体检测有助于两者的鉴别诊断[6]。本文中的两例患者均为中年女性(其中1例有家族史,其儿子为系统性红斑狼疮患者),急性起病,临床症状为肢体麻木无力、感觉异常(发痒)、尿便障碍等脊髓受损表现,无视力改变,颈胸腰段MRI 检查发现超过三个连续脊髓节段的异常信号,血清和脑脊液抗水通道蛋白4(AQP-4 )抗体阳性,支持视神经脊髓炎谱系疾病诊断。
系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种累及全身多脏器的自身免疫性的结缔组织病,主要累及皮肤粘膜、肾脏、骨骼肌肉及中枢神经系统,血清中可以检测到多种自身抗体。SLE患者中脊髓炎发病率估计在1%~3%[7],多在急性期或终末期出现,少数也可以作为首发症状,表现为脊髓炎或脑炎,Birnbaum等[8]将SLE伴发的脊髓炎分为灰质型和白质型,白质型大多开始症状较轻,病程易复发,缓慢进展,首发时肌力轻度下降,在非活动期或低活动期出现,脑脊液检查炎性反应不明显,MRI检查在T2像可发现异常信号,治疗效果相对较好。
视神经谱系疾病与系统性红斑狼疮均为自身免疫性疾病,两者常共存,推测一些潜在的机制可以解释NMOSD和SLE的结合:遗传和(或)环境因素可能使患者易获得自身免疫性疾病。系统性红斑狼疮可以使NMOSD的某些发病机制易化,炎症机制损害血脑屏障,使AQP4抗体易于通过受损的血脑屏障进入中枢神经系统,从而产生免疫反应。一些系统性风湿性疾病可以导致一些常见的免疫病理学改变,如血管病变[7]。
一些系统性红斑狼疮患者症状轻、起病隐匿、临床表现不典型,甚至在其他疾病的诊治中发现。本文两例患者均以脊髓炎表现起病,无典型的狼疮症状,但抗SS-A抗体,抗Ro-52抗体,抗SS-B抗体,抗核小体抗体均为阳性,诊断系统性红斑狼疮成立。所以在临床工作中,对疑似视神经脊髓谱系疾病的患者,即使没有SLE的临床表现和体征,临床医生也应考虑是否合并其他自身免疫性疾病,进一步完善免疫相关检查,以明确患者是否存在自身免疫性疾病。关于视神经脊髓谱系疾病与系统性红斑狼疮是相互叠加还是独立疾病,目前学界观点不一,但较多观点认为:对于AQP4抗体阳性伴发视神经炎、长节段横贯脊髓炎或脊髓受累的系统性红斑狼疮患者,应该按照视神经脊髓炎的诊治指南给予治疗[9]。本文两例患者在明确诊断后及时给予激素冲击或免疫治疗,症状体征明显减轻,复查脊髓MRI显示病灶明显缩小,治疗效果显著。
随着免疫学的发展,自身免疫性疾病如系统性红斑狼疮的诊断越来越规范,而视神经谱系疾病并非神经内科常见病,合并系统性红斑狼疮在临床上也相对罕见。在临床工作中,对怀疑视神经谱系疾病的患者在详细询问病史、仔细查体的前提下,应尽可能完善抗核抗体谱等风湿免疫指标,筛查自身免疫性疾病;反之,如果自身免疫病患者出现视力改变或肢体麻无力等脊髓症状时,也要及时检查影像学如头部和(或)脊髓MRI、视觉诱发电位等,了解是否合并NMOSD,减少漏诊、误诊。由于自身免疫性疾病有反复发作的倾向,诊治不当将会对患者本人及其家庭造成很大的负担,因此提高临床医生对该病的认识,有助于早期诊断和治疗,并对预防疾病的复发有积极的作用。
[参考文献]
[1]郎文娟,王 颖,朱明勤,等. 视神经脊髓炎谱系疾病合并类风湿性关节炎1例报告[J]. 中风与神经疾病杂志,2016,33(3):269-270.
[2]Wingerchuk DM,Lennon VA,Lucchinetti CF,et al. The spectrum of neuromyelitis optica[J]. Lancet Neurol,2007,6(9):805-815.
[3]Wingerchuk DM,Brenda B,Bennelt JL. International consensus diagnostic criberia for neuromyelitis optica spectrum disorders[J]. Neurology,2015,85:177-189.
[4]Sellner J,Boggild M,Clanet M,et al. EFNS guidelines on diagnosis and management of neuromyelitis optica[J]. Eur J Neurol,2010,17( 8):1019-1032.
[5]尹 周,应鸣翅,李小平,等. AQP4抗体测定对中国人群NMO及HR-NMO临床意义的系统分析[J]. 中风与神经疾病杂志,2015,32(8):715-718.
[6]何 洋,刘广志,高旭光. 水通道蛋白4抗体与神经系统自身免疫病[J]. 中华内科杂志,2011,50(3):264-266.
[7]岳孟龙,刘沛东,刘洪波,等. 系统性红斑狼疮合并视神经脊髓2例及文献复习[J]. 郑州大学学报( 医学版),2014,49(3):422-425.
[8]Birnbaum J,Petri M,Thompson R,et al. District subtypes of myelitis in systemic lupus erythematosus[J]. Arthritis Rheum,2009,60(11):3378.
[9]徐锦锦,张海宁,王 雪,等. 视神经脊髓炎谱系疾病合并干燥综合征1例报告[J]. 中风与神经疾病杂志,2015,32(6):554-556.
猜你喜欢
艺术品鉴(2022年16期)2022-07-09
临床误诊误治(2021年12期)2021-12-04
临床误诊误治(2021年12期)2021-12-04
现代临床医学(2021年5期)2021-11-02
昆明医科大学学报(2021年4期)2021-07-23
北方论丛(2021年2期)2021-05-22
景德镇陶瓷(2021年1期)2021-03-24
保健文汇(2020年2期)2020-08-24
当代陕西(2019年19期)2019-11-23
作文评点报·作文素材初中版(2019年37期)2019-11-16
