
金红石TiO2纳米团簇与铀酰相互作用的相对论密度泛函理论计算
2018-05-05 06:22:40张红星袁福龙潘清江
无机化学学报 2018年5期
郑 明 张红星 袁福龙*, 潘清江*,
(1黑龙江大学功能无机材料化学教育部重点实验室,化学化工与材料学院,哈尔滨 150080)
(2吉林大学理论化学研究所,长春 130023)
0 引 言
自从核燃料在军事和工业应用以来,其引发的环境污染问题持续引人关注。为解决已造成的环境污染、存储已生成的高放核废料以及为将来长期安全利用核资源,矿物表面和锕系元素相互作用的界面化学研究凸显重要性[1-3]。在世界范围内,铀是一种常见的污染物,特别是在核废物管理设施、铀矿开采工厂以及重工业场地中。其中六价氧化态的铀酰离子是一种最为稳定的结构单元。由于铀酰在水中有很高的溶解度,所以铀酰可以借助流动的水进行迁移,进而不可避免地与生物接触[4]。由于铀元素具有特殊毒性、放射性及其处理成本昂贵等特点,为人类长期健康以及避免铀污染土壤、沉积物、地下水,需要我们深入了解铀,包括矿物/水界面吸附,的多种表面运输过程。研究铀酰离子与多种矿物质表面的吸附行为有助于对核污染铀的扩散进行评估;另外,有实验表明铀酰卤化物具有光催化作用[5]。将铀酰离子吸附在稳定的矿物质表面有利于解决核废料的回收和处理问题,也可以进一步研究铀酰离子的光催化性质[6-7]。
溶解的铀酰化合物吸附到矿物表面的过程在调控铀酰在地下水中迁移起着重要的作用[4]。这个过程涉及到一系列的界面物理/化学反应。还有许多复杂的因素,例如,pH值、金属离子、基底的表面性质(酸度和表面位点密度)和溶液的离子强度[1]。目前,光谱技术广泛用于研究铀酰与矿物的相互作用[8-10]。例如X射线吸收精细结构(EXAFS),能够得到TiO2吸附铀酰的结构、组成和铀酰的吸附模式。此外,还有一些铀酰吸附到其它矿物质的实验报道,包括赤铁矿、三水铝矿、高岭石、和蒙脱石,这也进一步丰富了矿物表面吸附锕系元素的界面化学。
除光谱技术外,理论计算是也是一种优选的探索铀酰与矿物表面相互作用的重要工具。它也成功地补充和支持了实验结果。如,Roques和Simoni课题组使用平面波密度泛函理论 (周期性边界条件PBC-DFT)计算铀酰金红石(110)在3个不同的吸附位点的结构和稳定性[11]。随后,我们课题组曾使用PBC-DFT计算多种铀酰吸附质与金红石(110)的吸附能力[12]。此外,也有很多关于铀酰吸附到α-Al2O3(001)、γ-Al2O3(100)/(110)、α-SiO2(001)、蒙脱石、高岭石(001)、碳化钛、刚玉 表面等方面的理论计算报道。与大多数使用PBC-DFT研究铀酰和无限表面的相互作用相比,采用适当尺度的纳米离子团簇(有限表面)作吸附剂模型的量子理论研究仍然很少[13]。
本文中,采用全电子相对论密度泛函理论方法计算金红石型TiO2纳米团簇对铀酰的吸附行为。考察金红石团簇层数和表面大小对吸附复合物结构、吸附作用能的影响,为探索矿物表面吸附锕系元素的界面行为、阐明它们结构与性质关系以及为锕系元素分离化学提供可参考理论依据。
1 计算细节
金红石型TiO2具有层状结构,以往的周期性边界条件研究通常采用4~6层、且中间层冻结的模型。另外,实验[8,14]和理论[11]研究在水合金红石表面发现3种铀酰化合物的吸附位点:bb(铀酰与TiO2表面的2个相邻桥连氧原子Ob在赤道方向配位)、bt(铀酰与TiO2表面的1个Ob和1个端基氧Ot配位)和tt(铀酰与TiO2表面的2个Ot配位)。研究表明具有bb位点的复合物最稳定,其次是具有bt和tt位点的化合物。因此,我们的模型使用bb吸附这一稳定结构模型。
所有结构优化使用Priroda程序[15]完成,并且没有任何对称性限制条件。采用全电子(AE)标量相对论方法、广义梯度近似(GGA)的PBE泛函以及全电子高斯基组(标记为B-I)。在优化结构基础上的频率计算没有发现任何虚频,确认其稳定结构本质。另外,还计算得到配合物的多种热力学参数和Mayer键级。
使用ADF2014程序[16],(1)计算反应物和产物的溶剂化能,得出在水溶液中的反应能;(2)给出了复合物在溶液中的电子结构信息;(3)对TiO2纳米粒子与铀酰的相互作用(U-Osurf)进行能量分解计算;(4)对复合物在 U-X(X=Osurf、OH2和 Oyl)键临界点(Bond critical points,BCPs)进 行 QTAIM[17](Quantum Theory of Atoms in Molecule)拓扑分析。计算使用6.0×6.0×6.0的积分格点参数。采用COSMO模型模拟溶剂化效应[18],其中溶剂水的介电常数为78.39。Klamt半径:金属原子(rU=0.170 nm和rTi=0.210 nm)和主族原子 (rO=0.172 nm、rC=0.200 nm和rH=0.130 nm)。计算使用标量相对论ZORA方法[19]、GGA-PBE泛函和Slater型TZP基组。在ZORA-TZP模型下,进行冻结核轨道处理:U的1s-4f轨道、Ti的1s-2p轨道和C/N/O的1s轨道;因此,对U的32个价电子 (5s25p65d106s26p65f36d17s2)和Ti的12个价电子(3s23p64s23d2)进行全电子计算。
此外,使用Gaussian09程序[20],结合Multiwfn 3.3.3程序[21],对复合物在U-X(X=Osurf,OH2和Oyl)键临界点进行更为全面的QTAIM分析,包括电子密度ρ(r)、拉普拉斯密度▽2ρ(r)、能量密度 H(r)和离域化指数δ。计算采用Stuttgart RSC 1997 ECP小核有效核势相对论方法和PBE泛函,对铀原子采用相应的赝势基组,而对其他原子(H、O和Ti)采用6-31G*基组。
2 结果与讨论
2.1 不同层TiO2纳米团簇吸附水合铀酰复合物的几何结构
为寻找能够合理描述金红石纳米粒子性质的金红石型TiO2纳米团簇模型,设计并优化1到4层4种不同层纳米团簇模型,表面积均为1.1 nm×0.6 nm,团簇模型具体分子式见表1;同时,优化这些团簇吸附水合铀酰的复合物结构,图1可以看出,TiO2纳米团簇采用bb位点吸附水合铀酰,而3个水分子在铀酰离子赤道进行饱和配位。一方面,这种铀酰离子赤道方向的五重配位特征证明了EXAFS光谱对TiO2吸附铀酰的结构测定[8,14];另一方面,我们的计算结果也与以前的理论计算[22]与实验合成的铀酰配合物结构[23]相符合。

图1 优化的不同层金红石型TiO2纳米团簇模型吸附水合铀酰复合物Fig.1 Structures of optimized aquo uranyl-adsorbed nanoparticle clusters(NPCs)of rutile TiO2with 1 to 4 layers
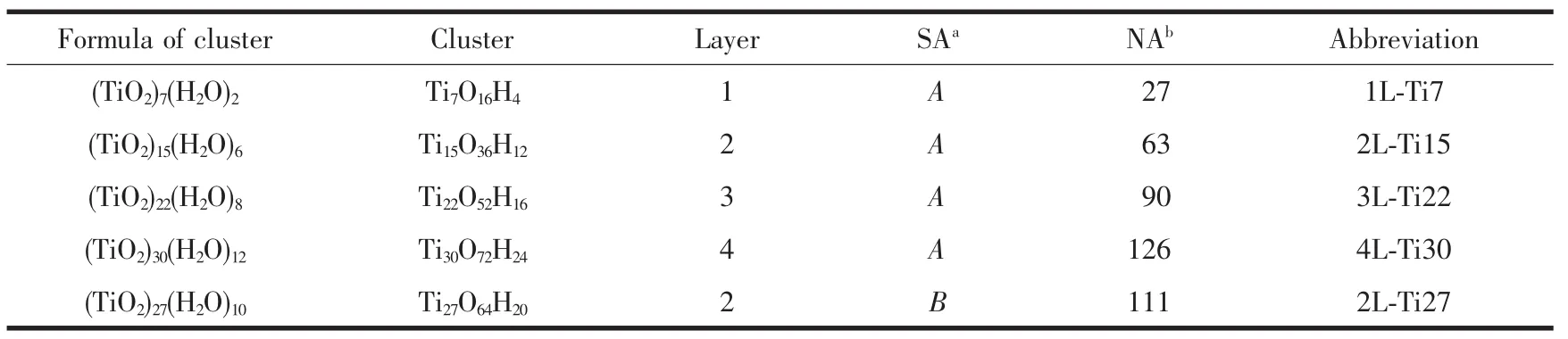
表1 不同层、不同表面积金红石型TiO2纳米团簇分析Table 1 Investigated rutile NPCs with different layers and surface areas
不同层TiO2纳米团簇模型吸附水合铀酰复合物的结构参数在表2中列出,4种不同层复合物的U=O的键长平均为0.180 nm,对比得到的不同层复合物之间U=O键键长,发现它们相差不到1%;与[UO2(H2O)5]2+和[UO2(H2O)3]2+的 U=O 键对比(分别为0.177 6和0.176 2 nm)(表S1),发现吸附在团簇模型上的铀酰双齿结构U=O键均比其稍长一些。
我们优化得到的水合铀酰U-OH2键键长在不同层复合物上有所不同,与[UO2(H2O)5]2+的U-OH2键对比(键长为0.248 6 nm),2层团簇模型上吸附的水合铀酰U-OH2键与其最为接近。优化得到U-Osurf键长0.233~0.238 nm,这一距离在已发现铀酰基配合物U-O距离范围内。与实验值0.233 nm[24]相比较,2层与4层复合物与实验值更为接近。优化得到的2层复合物的U-Ti间距与最近的MD结果0.24 nm[25]十分接近,比实验值0.305 nm稍长。这表明理论模型(cluster和PBC)与实验有一定的偏差。
4种不同层复合物的O=U=O角度在 156°~169°之间,O=U-Ti的角度在 89°~105°之间, 反映出2个-yl氧原子是背离团簇模型基底表面的。4种不同层复合物的U=O键的键级为2.41(平均值)反应出其具有三键的性质。1层到4层4种复合物铀酰赤道方面配位的U-Osurf的键级(平均值)(表3)分别为0.47、0.68、0.77、0.64,这足以表明铀酰和 4 种纳米团簇模型的每一个表面桥氧原子都形成单键,表现出强烈的吸附作用。
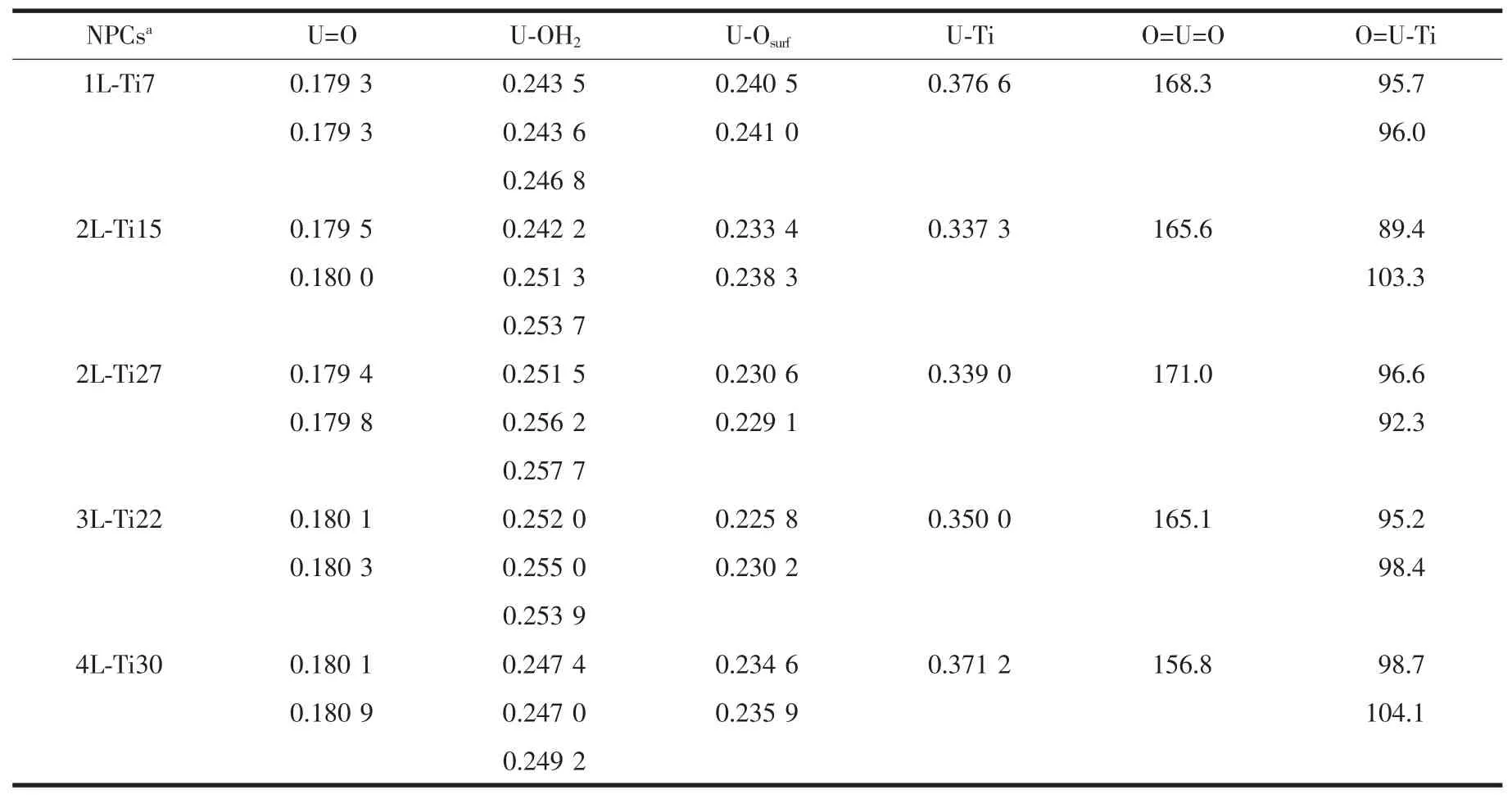
表2 优化的不同层金红石型TiO2纳米团簇吸附水合铀酰复合物的几何参数Table 2 Optimized geometry parameters of aquo uranyl adsorbed on NPCs with different layers(Distances in nm and angles in°)

表3 不同层金红石型TiO2纳米团簇吸附水合铀酰复合物的键级Table 3 Calculated bond orders of aquo uranyl adsorbed on rutile NPCs with different layers
2.2 不同层金红石型TiO2纳米团簇与水合铀酰的相互作用
使用Priroda程序,我们计算了从1层到4层的金红石型TiO2纳米团簇模型与水合铀酰复合物的形成反应能,研究TiO2吸附水合铀酰的热力学可能性。ΔrE、ΔrE0和ΔrG都是在气态条件计算的。使用ADF程序进一步计算在水溶液条件下额外的溶剂化作用ΔrG(sol)。设计反应如下:

计算得到所有的能量数据在表4中列出,基于每一个反应物和产物优化后的几何结构。铀酰离子在矿物表面的吸附受到pH值、铀酰离子浓度、吸附方式以及其他离子干扰等多方面因素的影响,我们这里只讨论纳米团簇模型层数不同导致反应能之间的差异。从图2中我们可以看到4种复合物ΔrE、ΔrE0、ΔrG的变化趋势相同。这样的能量变化趋势,是由于在1层和3层模型中水合铀酰中的水与模型的氧原子形成分子内氢键,从而使其形成反应能降低;4种复合物的ΔrE数值分别为-1.19、-2.63、-1.62、-2.38 eV,4种模型吸附水合铀酰的过程都是放热过程,并且2层和4层模型明显对铀酰具有更强的吸附作用;考虑到溶剂化效应的影响,水溶液中水的熵修正,4种模型的吸附ΔrG(sol)值计算出分别是 0.73、0.16、0.92、1.90 eV, 反应需要少许能量;从图2所示,ΔrG(sol)的趋势变化与ΔrE相比有所不同,是由于4层模型层数多、端基羟基较多,溶剂化效应对其影响较大。
同时为进一步研究铀酰的吸附作用,我们用ADF程序计算了水合铀酰吸附到4种不同层模型表面的相互作用能。计算公式如下:

表达式中ΔE为相互作用能 (IE),ELU-NPC是优化的水合铀酰吸附到4种不同层模型表面的化合物能量,ELU和ENPC分别是优化的吸附物-水合铀酰,吸附质-TiO2纳米团簇模型的能量。ADF程序计算出相互作用能ΔE的能量分解,更有助我们去了解铀酰吸附到TiO2的本质。

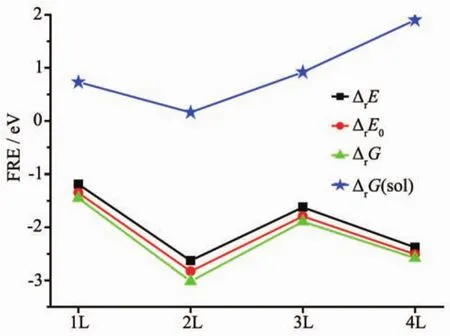
图2 在气态和水溶液条件下不同层金红石型TiO2纳米团簇吸附水合铀酰的反应能Fig.2 Formation reaction energies(FRE,eV)of aquo uranyl adsorbed on rutile NPCs with different layers in the gas phase and aqueous solution

表4 在气态和水溶液下不同层金红石型TiO2纳米团簇吸附水合铀酰的反应能aTable 4 Calculated formation reactionaenergies of aquo uranyl adsorbed on rutile NPCs model with different layers in the gas phase and aqueous solution
在上面的式子中,EElectro,EPauli,EOrbit和 ESteric分别表示静电相互作用能,Pauli排斥能,轨道相互作用能,以及空间相互作用能。EKinetic,ECoulm和EXC分别表示动力学能,库仑能和交换相关能。相互作用能ΔE与能量分解的数据在表5中列出。水合铀酰吸附到1层到4层模型的相互作用能ΔE分别是-5.80、-8.07、-8.35、-9.29 eV。更负的相互作用能意味着吸附物和基底之间更强的相互作用;对不同层模型的相互作用能通过式(4)分解,轨道相互作用能在4种不同层数模型与铀酰的吸附过程中占据主导地位,增加模型层数显著增强了模型吸附水合铀酰的能力;从能量分解数据来看,随模型层数的增加模型的原子数量增多,静电相互作用能也随之增加。与其他模型相比,4层模型的体积大、原子数多,其空间相互作用能(-1.24 eV)相对较大,其静电吸引(-8.23 eV)大于 Pauli排斥能(6.99 eV)。

表5 ADF程序计算的水合铀酰与不同层TiO2纳米团簇模型的相互作用能的分解数据Table 5 Decomposition of the interaction energy between aquo uranyl and rutile NPCs model with different layers Calculated by the ADF Code
2.3 表面积对2层金红石型TiO2纳米团簇模型吸附水合铀酰复合物结构和性质的影响
为了进一步探索表面积对2层金红石型TiO2纳米团簇模型吸附水合铀酰复合物的影响,我们另外选取表面积更大的模型进行比较研究。模型2LTi27的表面积为1.1 nm×1.2 nm(2L-Ti15为1.1 nm×0.6 nm),分子式为(TiO2)27(H2O)10。我们采用Priroda程序在气相条件下对模型进行优化,同时优化吸附水合铀酰复合物的结构,水合铀酰采用bb位点吸附在模型表面上。优化的相应结构见图3,选定的几何参数在表2中列出。从键长、键角几何参数看,2种模型之间的差异很小,表面积的改变对于复合物的整体结构来说没有明显的影响。

图3 优化不同表面积的2层金红石型TiO2纳米团簇吸附水合铀酰的几何结构Fig.3 Structures of optimized aquo uranyl-adsorbed 2-layer rutile NPCs with different surface areas
同时我们还计算了这2种2层金红石型纳米团簇模型吸附水合铀酰复合物的形成反应能和相互作用能,用Priroda程序在气态条件计算ΔrE、ΔrE0、ΔrG。使用ADF程序进一步计算在水溶液条件下额外的溶剂化作用ΔG(sol)以及相互作用能,能量数据分别在表4表5中列出。2种复合物的ΔrE数值分别为-2.63、-2.71 eV,形成复合物都是放热过程。从图3中可以看出2L-Ti27模型复合物的热力学函数数值整体高于2L-Ti15模型复合物,但2种复合物的热力学函数之间相差很小。2种模型与水合铀酰相互作用能ΔE为-8.07和-8.19 eV,与水合铀酰之间具有很强的相互作用。能量分解发现,2种模型和铀酰的化学键作用为轨道相互作用主导的,2L-Ti27模型轨道相互作用占98.8%,2L-Ti15模型轨道相互作用占93.9%。
从结构和能量计算对比,表面积对2层金红石型TiO2纳米团簇模型影响很小。我们确定2层、表面积为1.1 nm×0.6 nm、包括63个原子的纳米团簇模型2L-Ti15能够合理描述金红石纳米粒子性质。
2.4 2L-Ti15模型吸附水合铀酰复合物的U-Osurf键性质分析及电子结构
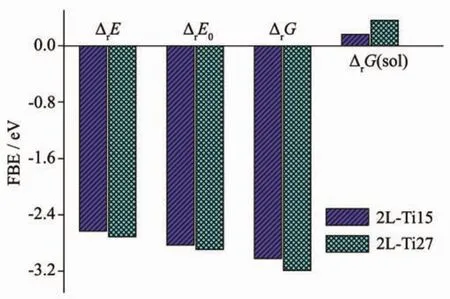
图4 在气态和水溶液条件下不同表面积的2层金红石型TiO2纳米团簇吸附水合铀酰的反应能Fig.4 Formation reaction energies in the gas phase and aqueous solution with aquo uranyl-adsorbed 2-layer NPCs with different surface areas
水合铀酰吸附到2L-Ti15模型采用bb位点进行吸附,水合铀酰与底物TiO2纳米团簇模型通过U-Osurf键直接相连。为了更清楚的分析U-Osurf键,我们对其进行QTAIM分析。在QTAIM分析中,2个成键的原子之间,键的临界点(BCP)是键径上电子密度最低点。临界点参数例如电子密度ρ(r)、拉普拉斯电子密度▽2ρ和能量密度H(r)经常用来表征化学键的性质和强度。ρ(r)>0.20 a.u.,▽2ρ<0,表明为共价键或开壳层作用,ρ(r)<0.10 a.u.,▽2ρ>0,说明为闭壳层作用(离子键、范德华作用或氢键等)。椭率ε值接近于零表明是单键或者三键,当达到ca.0.45时表明双键特征。离域化指数δ值被视为测量2个成键原子间的键级。
计算的U-Osurf键、U=O键、U-OH2键的临界点BCP参数在表6中列出。对于2个U-Osurf键的能量密度H(r)都为负值且绝对值较小,表明U-Osurf之间的键具有共价性且强度较弱;电子密度ρ(r)值都很小,分别为 0.072 7、0.066 9 a.u.,并且▽2ρ都为正值分别为0.277 1、0.240 9 a.u.,表明U-Osur键有闭壳层作用(趋于离子键),即显示出极性。我们指认UOsurf键为介于离子和共价之间的配位键。计算出2个U-Osurf键的椭率ε较小,离域化指数δ(U,O)值分别为0.504 0、0.502 4,这表明U与表面Osurf之间的键为单键,另一方面键级数据(表3)也可以证明它们之间成键为单键。计算U-OH2键的临界点BCP参数与U-Osurf键参数相近,U与H2O之间的键为单键,但由于其H(r)绝对值相对U-Osurf键的H(r)绝对值较小,其共价作用相对较弱。U=O键可以通过其椭率ε值0.017 0,0.007 6以及离域化指数δ(U,O)2.018 0,1.975 3判断成键为三键。同时我们用ADF程序也计算了它们临界点处电子密度ρ(r),拉普拉斯电子密度(▽2ρ),对比发现它们之间的差别不大。
为了进一步了解复合物的电子性质,我们用ADF程序计算了2L-Ti15模型吸附水合铀酰复合物的基态波函数,重要分子轨道 (MO)图和态密度(DOS)图分别见图5、图S4和图6。态密度分析对理解和掌握铀酰吸附机理具有重要作用。最高占据轨道(HOMO)和最低非占据轨道(LUMO)的能级分别是-7.285和-4.881 eV,能级差为2.404 eV。在图6中可以看到2L-Ti15金红石型TiO2纳米团簇模型吸附水合铀酰复合物的HOMO和HOMO-3主要由基底2p(Osurf)和少量铀酰fσ(U=O)组成,其HOMO-11主要是基底 2p(Osurf)成分。 fπ(U=O),dπ(U=O)轨道位于较低能量区域,2种fπ(U=O)分别位于H-49和H-54,dπ(U=O)轨道位于H-58和 H-67处。在态密度(DOS)图中可以看到铀酰离子周围的水配位体对高位占据轨道没有贡献,在H-11到H-49之间的部分轨道有少许贡献。水合铀酰离子的氧Oyl同样对高位占据轨道没有贡献,在低位占据轨道有少许贡献。
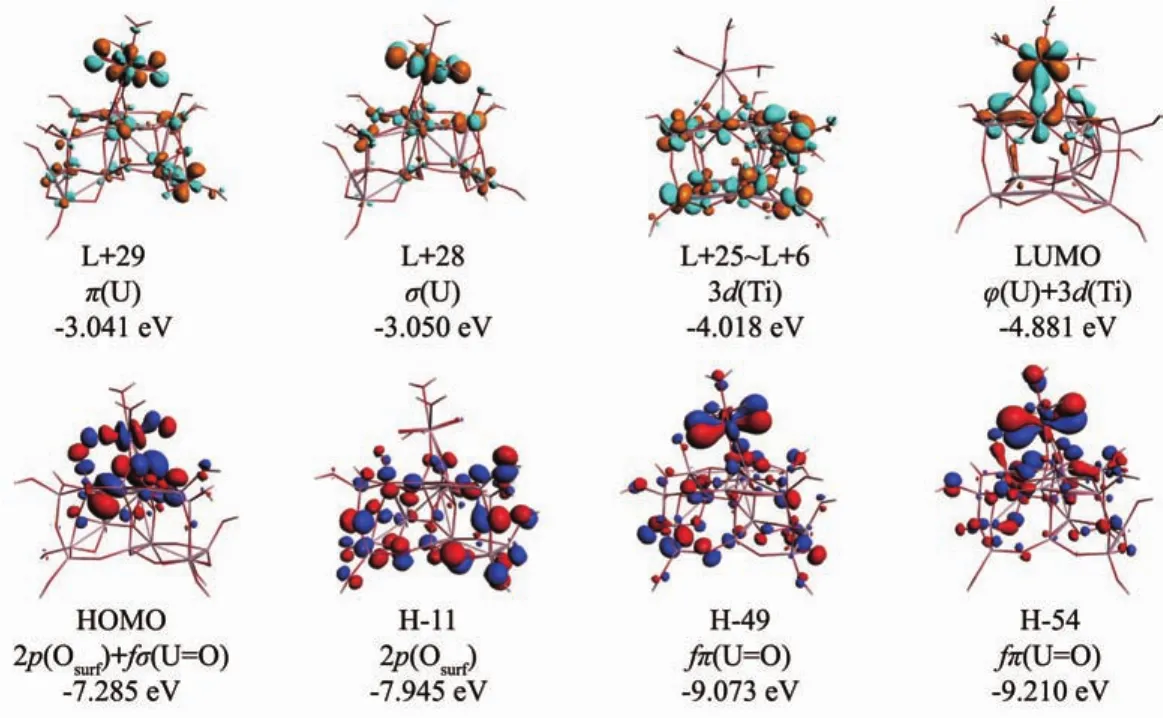
图5 选取2L-Ti15吸附水合铀酰复合物的部分轨道电子云图,其中具有相似性质的轨道仅绘制一个具有代表性的Fig.5 Selected orbitals of the aquo uranyl-adsorbed 2L-Ti15,where orbitals with similar character were shown with a representative one
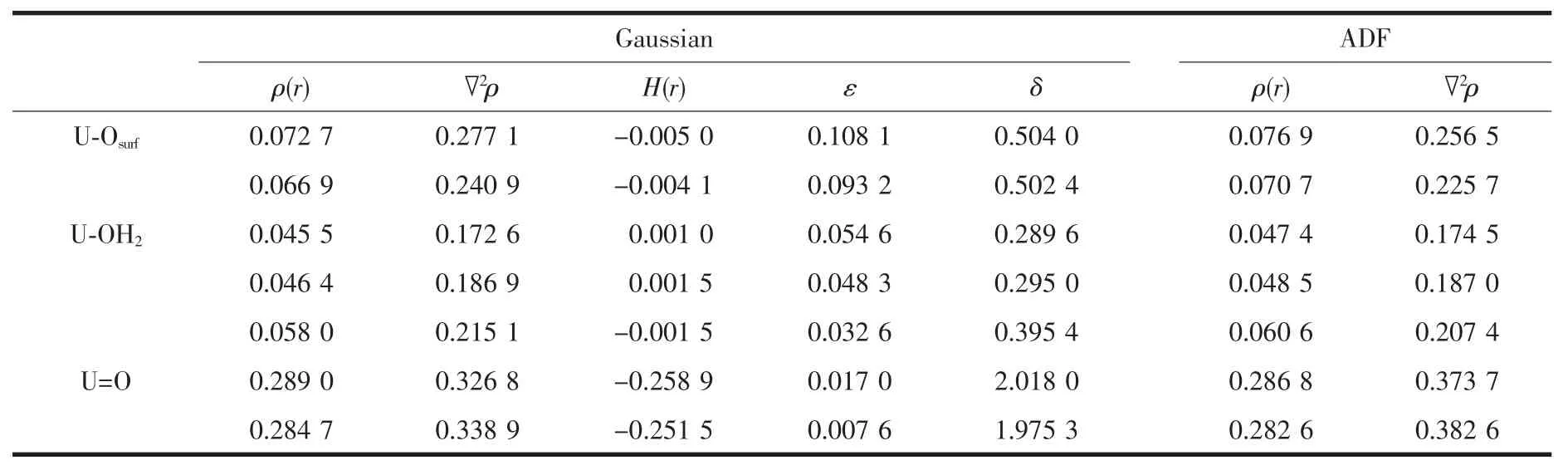
表6 计算的U-X键临界点处QTAIM参数(单位为a.u.)和离域化指数δTable 6 Calculated electron density[ρ(r)],Laplacian[▽2ρ(r)],energy density[Н(r)]and ellipticity(ε)at U-X bond critical points(BCPs)(all values in a.u.),together with the delocalization index δ

图6 复合物2L-Ti15-[UO2(H2O)3]2+的态密度图Fig.6 Density of states(DOS)of the 2L-Ti15-[UO2(H2O)3]2+complex
在该复合物中有5个5f(U)低位非占据轨道,LUMO-LUMO+4(L+4),一些3d(Ti)轨道混合到LUMO和L+4中。在高能量区域L+4以上,基底的3d(Ti)的形成最多未填充轨道范围是从L+6到L+25。由于5fπ(U)和 2pπ(Oyl)之间的相互排斥作用,由此产生的π*轨道(U=O)出现在能量更高的区域L+28和L+29处。为了与2L-Ti15模型吸附水合铀酰复合物对比,在支持信息中列出2L-Ti27模型吸附水合铀酰复合物的轨道图(Fig.S1)和态密度(DOS)图(Fig.S2)。可以看出,其能级差为2.07 eV,高位占据轨道(HOMOHOMO-34)主要是基底 2p(Osurf)成分,而铀酰 fσ(U=O)轨道转移到较低能量区域(H-35)。
3 结 论
本文采用全电子相对论密度泛函理论方法计算金红石型TiO2纳米团簇对铀酰的吸附行为。
设计1层到4层4种模型,考察金红石团簇层数和表面大小对吸附复合物结构、吸附作用能的影响。通过比较优化的结构参数和计算的吸附作用能,确定2层、表面积为1.1 nm×0.6 nm、包括63个原子的纳米团簇2L-Ti15能够合理描述金红石纳米粒子性质。
对2L-Ti15-[(UO2)(H2O)3]2+复合物结构优化和性质计算表明,在气相条件下,纳米团簇模型对铀酰的吸附反应为放热过程,能量为-3.02 eV;在引入溶剂化效应时,反应则需要吸收0.16 eV能量。就热力学能量而言,反应能够在较温和的条件下进行。这与实验报道的TiO2纳米粒子可以吸附铀的结果相符合。
计算表明纳米团簇和铀酰存在共价化学键作用。首先,优化得到U-Osurf键长0.233~0.238 nm;其次,对U-Osurf进行能量分解,发现这一作用是轨道相互作用主导的,而其它的静电吸引则略大于Pauli排斥;另外,QTAIM拓扑分析指认U-Osurf键为以介于离子和共价之间的配位键,其强度大于复合物中的U-OH2键,但却小于U=O作用。波函数分析表明复合物的HOMO-LUMO带隙为2.40 eV,相对吸附前纳米团簇半导体粒子的3.35 eV变窄;复合物的电子结构特征为HOMO轨道主要由TiO2的O(2p)贡献,并混有少量的σ(U=O)成键性质,而LUMO轨道则为U(5f)性质为主、Ti(3d)仅有少量修饰作用。
总之,我们的研究一方面为半导体材料吸附分离高毒性、高辐射性锕系元素提供理论支持。另一方面,从吸收光谱角度而言,由于复合物体系带隙变窄,使其在可见光区域具有更强的捕光性能;吸附在TiO2纳米粒子半导体表面的铀酰离子对光生电子有吸引作用、促进电子-空穴的空间分离、降低电子-空穴复合几率;因而有作为光催化剂应用的潜在可能性。
参考文献:
[1]Hashke J M,Stakebake J L.The Chemistry of the Actinide and Transactinide Elements.Dordrecht:Springer,2006:3199-3272
[2]Chen Z,Zhuang Z Y,Cao Q,et al.ACS Appl.Mater.Interfaces,2014,6(2):1301-1305
[3]GU Jia-Fang(辜家芳),XU Ke(许可),CHEN Wen-Kai(陈文凯).Chinese J.Inorg.Chem.(无机化学学报),2017,33(9):1579-1586
[4]Natrajan L S,Swinburne A N,Andrews M B,et al.Coord.Chem.Rev.,2014,266:171-193
[5]Sandhu S S,Kohli K B,Brar A S.Inorg.Chem.,1984,23(22):3609-3612
[6]Nieweg J A,Lemma K,Trewyn B G,et al.Inorg.Chem.,2005,44(16):5641-5648
[7]Krishna V,Kamble V S,Gupta N M,et al.J.Phys.Chem.C,2008,112(40):15832-15843
[8]Vandenborre J,Drot R,Simoni E.Inorg.Chem.,2007,46(4):1291-1296
[9]Zhao S,Zhong Y,Guo Y,et al.Acta Chim.Sinica,2016,74(8):683-688
[10]GU Jia-Fang(辜家芳),MAN Mei-Ling(满梅玲),LU Chun-Hai(陆春海),et al.Chinese.J.Inorg.Chem.(无机化学学报),2012,28(7):1324-1332
[11]Perron H,Domain C,Roques J,et al.Inorg.Chem.,2006,45(17):6568-6570
[12]Pan Q J,Odoh S O,Schreckenbach G,et al.Dalton Trans.,2012,41(29):8878-8885
[13]Zhao H B,Zheng M,Schreckenbach G,et al.Inorg.Chem.,2017,56(5):2763-2776
[14]Dossot M,Cremel S,Vandenborre J,et al.Langmuir,2006,22(1):140-147
[15]Laikov D N.Chem.Phys.Lett.,1997,281(1/2/3):151-156
[16]Fonseca Guerra C,Snijders J G,te Velde G,et al.Theor.Chem.Acc.,1998,99(6):391-403
[17]Bader R F W.J.Phys.Chem.A,1998,102:7314-7323
[18]Pye C C,Ziegler T.Theor.Chem.Acc.,1999,101(6):396-408
[19]van Lenthe E,Baerends E J,Snijders J G.J.Chem.Phys.,1993,99(6):4597-4610
[20]Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian09,Gaussian,Inc.,Wallingford CT,2009.
[21]Lu T,Chen F.J.Comput.Chem.,2012,33(5):580-592
[22]Pan Q J,Schreckenbach G.Inorg.Chem.,2010,49(14):6509-6517
[23]Arnold P L,Jones G M,Odoh S O,et al.Nat.Chem.,2012,4(3):221-227
[24]Den Auwer C,Drot R,Simoni E,et al.New J.Chem.,2003,27(3):648-655
[25]Sebbari K,Roques J,Simoni E,et al.Surf.Sci.,2012,606(15/16):1135-1141
猜你喜欢
原子与分子物理学报(2021年1期)2021-03-29 07:29:36
化学与生物工程(2021年2期)2021-02-25 12:56:46
——以金红石为例
中国金属通报(2020年23期)2020-03-15 07:54:30
无机材料学报(2020年2期)2020-03-08 14:05:54
分析化学(2019年3期)2019-03-30 10:59:24
高师理科学刊(2016年8期)2016-06-15 20:27:49
无机盐工业(2016年2期)2016-03-15 03:33:44
化工矿产地质(2015年3期)2015-12-04 02:09:43
核科学与工程(2015年2期)2015-09-26 11:57:21
食品工业科技(2014年13期)2014-03-11 18:17:12
