
鲁比前列酮的合成
2018-04-19 08:35游军辉刘建平杜祖银
山东化工 2018年6期
游军辉,曹 金,刘建平,余 俊,杜祖银
(江苏豪森医药集团有限公司,江苏 连云港 222000)
鲁比前列酮(Lubiprostone),化学名:(-)-7-[(2R,4aR,5R,7aR)-2-(1,1-二氟戊基)-2-羟基-6-氧代八氢环戊烷并[b]吡喃-5-基)庚酸。本品是由苏坎波公司研发的一种治疗便秘型肠易激综合征药物,美国FDA于2006年4月批准上市。本品为一种氯离子通道激活剂,主要适用慢性特发性便秘和便秘型肠易激综合征的治疗[1]。文献[2]采用先上含氟侧链,后上不含氟侧链的方法来制备鲁比前列酮,该工艺在上含氟侧链时使用了剧毒试剂乙醇铊和苯,不适合工业化生产;文献[3]采用先上不含氟侧链,后上含氟侧链的方法制备鲁比前列酮,该工艺最后一步采用钯碳氢化来脱去苄基保护基,该方法容易在成品中引入两个还原杂质1和2(结构如下),从而造成产品纯度低,提纯困难。
本研究参考相关文献[4-5],提供了一条新的鲁比前列酮的制备工艺,即以科立内酯二醇为原料,经羟基的选择性保护和氧化、维悌希反应、内酯还原等操作得到化合物10,然后经还原、氧化和磷酸水解得到鲁比前列酮。
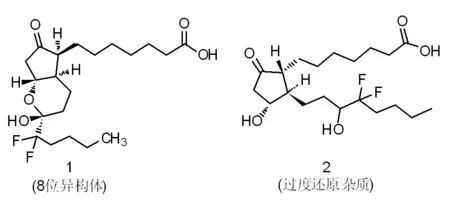
图1 鲁比前列酮中的过度还原杂质

图2 改进后的鲁比前列酮合成路线
1 仪器和试剂
Bruker AV500 核磁共振仪,CDCl3为溶剂,TMS为内标;Bruker AV200 核磁共振仪,CDCl3为溶剂,TMS为内标;Elementar Vario EL III型元素分析仪测定化合物中C、H含量;Nicolet FT IR-200红外光谱仪,KBr压片法;Agilent 6224飞行时间质谱仪,样品的甲醇溶液,范围:200~450 m/z;安捷伦1260液相色谱仪。
所有试剂均采购于国药集团化学试剂有限公司;戴斯马丁氧化剂购于瑞一医药科技有限公司,含量>98%;化合物3的合成参考文献[5]方法合成,化合物6和化合物9的合成参考文献[3]方法合成。
2 制备步骤
2.1 化合物4的合成
将205 g的四丁基氟化铵加入到105 g化合物3和1500 mL THF的混合液中,室温搅拌12h,将反应浓缩至无液滴流出后,加入硅胶制砂上样,用石油醚/乙酸乙酯(60∶1~20∶1)柱层析得化合物4 45.2 g油状产品。收率:62.2%。
2.2 化合物5的合成
将90 g戴斯马丁氧化剂加入到45g 化合物4和900 mL二氯甲烷的混合液中,室温反应12h,过滤,将滤液加入硅胶制砂过柱,用石油醚/乙酸乙酯(40∶1~10∶1)柱层析得化合物5 30.4 g油状产品。收率:68.1%。
2.3 化合物7的合成
氮气保护下,将化合物6 45 g溶于100mL MTBE中,加入一水氢氧化锂6.9 g室温反应1h。加入化合物5 30 g和200 mL 二氯甲烷的混合溶液,再加入8 mL H2O,反应体系升温到45℃反应24h。向体系加480 mL水,搅拌20min,乙酸乙酯提取三次,合并有机相,无水硫酸钠干燥,过滤,浓缩,用石油醚/乙酸乙酯(30∶1~5∶1)柱层析得化合物7 28 g油状产品。收率:61.4%。
2.4 化合物8的合成
氮气保护下,将27 g化合物7和230 mL甲苯冷至 -78℃。缓慢滴加248 mL 25%的DIBAL-H的正己烷溶液,加完后,保温反应2h。缓慢滴加407 mL 25%的酒石酸钾钠水溶液,加完后升温至室温,继续剧烈搅拌3h,至溶液澄清,分出有机相,水相用乙酸乙酯提取。合并所有有机相加入无水硫酸钠干燥,过滤,浓缩,用石油醚/乙酸乙酯(30∶1~1∶1)柱层析得化合物8 24 g油状产品。收率:88.0%。
2.5 化合物10的合成
将50 g 化合物9和125 mL THF冷至0度,搅拌10 min,滴加400 mL LHMDS的四氢呋喃溶液,加完保温,搅拌10~15 min后,滴加24 g 化合物8的300 mL四氢呋喃溶液,加完后保温反应0.5h。加入1000mL水和1000mL甲基叔丁基醚,室温搅拌20 min,分出水相,调pH值至6~7,用乙酸乙酯提取,合并有机相,无水硫酸钠干燥,减压浓缩,石油醚/乙酸乙酯(25∶1~10∶1)柱层析得25g化合物10。收率:85.7%。
2.6 化合物11的合成
将化合物10 14 g 溶于28 mL乙酸乙酯中,加入10% Pd/C(50% H2O) 5 g,抽换氢气,室温常压反应2 h,过滤,滤液减压浓缩得7-[(1R,2R,3R,5S)-2-(4,4-二氟-3-羟基辛基)-5-羟基-3-(2-四氢吡喃氧基)环戊基]庚酸(11) 14 g,白色固体。收率:99.2%。1HNMR (200 MHz,CDCl3):(ppm):4.71~4.58 (1H,m),4.18~3.96(2H, m),3.96~3.60(2H,m),3.60~3.42(1H,m),2.35(2H,t,J=7.6Hz),2.13~1.17(30H,m),0.93(3H, t,J=7.2Hz)。
2.7 化合物12的合成
将化合物11 12g 溶于240 mL二氯甲烷中,加入戴斯马丁氧化剂16g,室温反应14h,加饱和亚硫酸钠水溶液淬灭多余的戴斯马丁氧化剂,待基本澄清后加饱和碳酸氢钠水溶液30mL搅拌20 min,加乙酸乙酯萃取,合并有机相,无水硫酸钠干燥,过滤,浓缩,用石油醚/乙酸乙酯(20∶1~5∶1)柱层析得7-[(1R,2R,3R,5S)-2-(4,4-二氟-3-氧代辛基)-5-氧代-3-(2-四氢吡喃氧基)环戊基]庚酸(12) 10 g浅黄色油状物,收率:84.0%。1HNMR (200 MHz,CDCl3):δ (ppm):4.71~4.58 (1H, m),4.18~3.96(2H,m),3.96~3.60(2H,m),3.60~3.42(1H,m),2.35(2H,t,J=7.5 Hz),2.13~1.17(30H,m),0.93(3H, t,J=7.1Hz)。
2.8 鲁比前列酮的合成
将化合物12 10 g和50 mL乙腈冷至0~5℃,加入乙腈/85 %磷酸/水=10 mL/50 mL/10 mL的混合溶液,加完后保温反应2h,加水100 mL稀释,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,浓缩,用石油醚/乙酸乙酯柱(10∶1~3∶1)层析得鲁比前列酮粗品约8.1 g。将其用乙酸乙酯/正己烷析晶,干燥得7.57 g白色固体,收率:92%,HPLC:99.94 %。元素分析:C%实测值:61.74%、61.78%,计算值:61.52%;H%实测值:8.19%、8.17%,计算值:8.26%。ESI/HRMS(m/z):413.21013 [M+Na]+;IR(KBr,νcm-1):3383(m),2962(m),2932(s),2859(m),1731(s),1705(s),1427(w),1409(w),1377(w),1282(m),1232(m),1158(m),1067(m);1H NMR(500MHz,CDCl3)δ:0.94(t,3H),1.32~1.41(m,8H),1.47~1.68(m,7H),1.79~1.86 (m,3H),1.88~2.03(m,4H),2.22~2.28(m,1H),2.34(t,2H),2.54~2.59(m,1H),2.0~5.0(br,1H),4.15~4.21(m,1H),7.6~12.8(br,1H)。13C NMR(500MHz,CDCl3)δ:13.8,22.5,23.0,33.5,24.5,26.9,27.2,28.0,28.7,29.4,30.3,30.5,30.7,33.9,43.6,46.0, 53.1,71.6,97.0, 97.2,97.4,120.3,122.3,124.3,179.6.213.9。
3 结论
本研究采用先还原再氧化的操作,成功的克服了现有合成工艺中的缺点,有效降低了鲁比前列酮中的过度还原杂质的含量,是一条具有很好工业化前景的工艺路线。
[1] 白秋江,郑 敏.鲁比前列酮[J].中国新药杂志,2007,16 (13):1060-1061.
[2] Ryuzo Ueno,Ryuji Ueno,Ichie Kato,et al.Prostaglandins E and anti ulcers containing same:US,6265440[P].1998-05-06.
[3] Dino Alberico,Joshua Clayton,Boris Ivanovich Gorin,et al.Prostaglandin synthesis and intermediates for use therein:US,2010056807[P].2008-11-14.
[4] 赵育磊.Corey内脂二醇合成工艺的改进及其在前列素类药物合成中的应用[D].山东:曲阜师范大学,2012.
[5] 张富尧,高书三.一种制备鲁比前列酮的中间体、其制备方法以及通过其制备鲁比前列酮的方法:CN,103787942[P].2014-05-14.
猜你喜欢
硫酸工业(2020年2期)2020-04-16
吉林农业(2019年6期)2019-06-11
教育教学论坛(2018年38期)2018-09-25
中成药(2017年10期)2017-11-16
今日重庆(2017年6期)2017-07-05
中成药(2017年4期)2017-05-17
动物营养学报(2015年9期)2016-01-07
中国洗涤用品工业(2015年4期)2015-02-28
浙江畜牧兽医(2014年5期)2014-02-27
小小说月刊(2009年12期)2009-05-14
