
HPLC-DAD 法检测盐酸左旋咪唑注射液中左旋咪唑的含量*
2018-04-16 02:35林仙军章荣叶周芷锦汤赛飞王彬陈晓林陆春波
中国动物保健 2018年12期
林仙军,章荣叶,周芷锦,汤赛飞,王彬,陈晓林,陆春波
(1.浙江省兽药饲料监察所 杭州 311101;2.台州市屠宰管理所 浙江台州 318000;3.浙江建安检测研究院有限公司 杭州 310006)
盐酸左旋咪唑是广谱驱虫药和生物反应调节药[1],对牛、羊、猪、犬、猫和禽的胃肠道线虫、肺线虫及猪肾虫病具有良好驱虫活性[2-3]。盐酸左旋咪唑注射液质量标准收载于《中国兽药典》2015 年版一部[4],标准采用三氯甲烷萃取、高氯酸滴定法(非水测定法)进行含量测定,方法操作繁琐、费时,高氯酸滴定液能与多种药物反应,专属性不高。检测中所用试剂三氯甲烷、醋酐等对人体有危害性,对环境污染也较严重。
目前文献报道测定盐酸左旋咪唑含量的方法除高氯酸滴定法外,主要以高效液相色谱法为主,有测定饲料[5]、绵羊血浆[6]、猪鸡组织[7]、盐酸左旋咪唑粉[8]、复方盐酸左旋咪唑注射液[9]和万乳康[10]等各类样品中盐酸左旋咪唑的含量,以及盐酸左旋咪唑原料中的有关物质[11],但目前尚未见高效液相色谱法(HPLC)测定盐酸左旋咪唑注射液中左旋咪唑含量的报道。因此,本研究对完善盐酸左旋咪唑注射液质量标准有促进作用。
1 材料与方法
1.1 仪器设备
Agilent 1260 高效液相色谱仪,配DAD 检测器(美国Agilent 公司);AG-285 电子天平(Mettler公司);KQ-500E 型超声波清洗器(昆山市超声仪器有限公司);METTLER TOLEDO 320 酸度计(Mettler 公司)。
1.2 试剂与药品
乙腈、甲醇均为色谱纯,德国默克公司;磷酸、三乙胺、盐酸、过氧化氢和氢氧化钠均为分析纯;水为超纯水。左旋咪唑对照品来源为中国食品药品检定研究 院, 批 号100167-201203,含量99.9%。盐酸左旋咪唑注射液来源为浙江凯特道格药业有限 公 司, 批 号 为20180901、20180902 和20180903,规 格 为10m L ∶0.25g。
1.3 对照品溶液和供试品溶液的配制
称取左旋咪唑对照品适量,精密称量,用流动相超声溶解并定量稀释制成每1mL 中约含100µg 的溶液,摇匀,作为对照品溶液;精密量取供试品适量,用流动相超声溶解并定量稀释制成每1mL 中约含100µg 的溶液,摇匀,作为供试品溶液。
1.4 标准曲线系列溶液的配制
取左旋咪唑对照品适量,用流动相超声溶解并稀释制成每1m L 中含左旋咪唑500µg 的溶液,摇匀,作为对照品溶液。取对照品溶液适量,用流动相稀释成浓度分别为1、5、20、50 和100µg/m L 的溶液。
1.5 高效液相色谱条件和系统适应性试验
色 谱 柱:Agilent Eclipse XDB-C18,内径4.6mm,柱长250mm,粒径5μm;用二极管阵列检测器进行扫描,扫描范围为190~400nm, 记 录212nm 波 长处的色谱图,流速:1.0m L/min;进样量:10μL;柱温:30 ℃;流动相为0.05mol/L 磷酸二氢钾溶液(三乙胺调节p H 值至7.0)-乙腈(80 ∶20,v/v)。
1.6 非水滴定的方法
精密量取本品适量(约相当于盐酸左旋咪唑0.25g),置分液漏斗中,加氢氧化钠试液5m L,稍振摇后,精密加入三氯甲烷50m L,振摇提取。静置分层后,精密量取三氯甲烷液25m L,加冰醋酸15m L、醋酐2m L 与结晶紫指示液1 滴,用高氯酸滴定液(0.1mol/L)滴定至溶液显蓝色,并将滴定的结果用空白试验校正。每1m L高氯酸滴定液(0.1mol/L)相当于24.08mg 的C11H12N2S·HCl。
2 结果
2.1 标准曲线及线性关系考察
取1.4 项的溶液,按照建立的色谱条件进样检测,得到左旋咪唑对照品溶液色谱图见图1,光谱图见图2。以系列对照品溶液浓度为横坐标,峰面积为纵坐标绘制标准曲线,左旋咪唑在1~500µg/m L 浓度范围内线性关系良好,线性方程Y=44.190X+17.116,相关系数r=0.999 99。
2.2 稳定性试验
取批号为20180901 的左旋咪唑供试品溶液,分别于0、2、4、6、12 和24h 进行测定,测得左旋咪唑峰面积为RSD 为1.0%,表明供试品溶液稳定性良好。
2.3 HPLC-DAD法和非水法比较
分别用HPLC 法和非水法对实际样品进行测定,数据见表1。实际样品色谱图见图3,光谱图见图4。左旋咪唑的含量与非水测定法一致,相对偏差在1.0%以内。
2.4 准确度
按照盐酸左旋咪唑注射液配制方法,分别按2.5、5.0 和10.0mg/mL 三个浓度添加盐酸左旋咪唑原料,每个浓度分别制备5 个平行样品,求得添加回收率及相对标准偏差,测定结果见表2。

图1 左旋咪唑对照品溶液

图2 左旋咪唑对照品溶液扫描光谱图
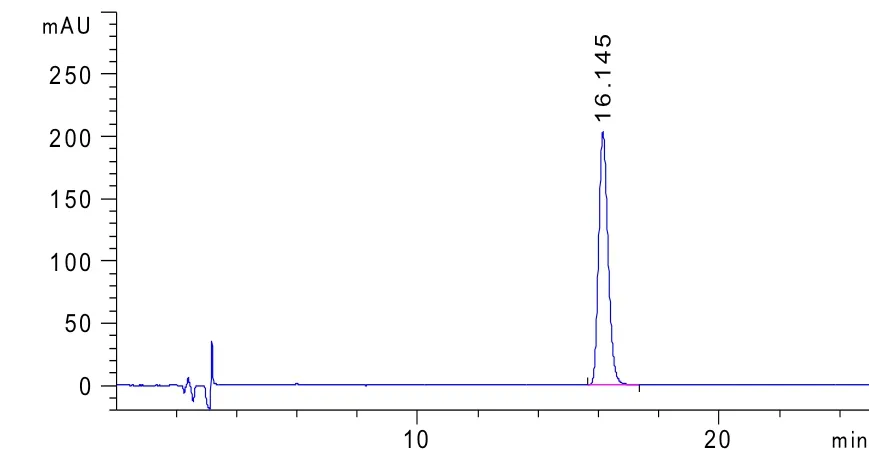
图3 左旋咪唑注射液供试品色谱图(批号20180901)
3 讨论和小结
1) 取标准曲线系列溶液中100µg/mL 对 照 品 溶 液, 在190~400nm 波长范围内进行DAD扫描。结果左旋咪唑在212 nm 的波长处有最大吸收,见图2。因此,选择212 nm 作为检测波长。
2)用乙腈-水作为流动相时,左旋咪唑色谱峰拖尾较为严重。在流动相中加入三乙胺、磷酸、磷酸二氢钾溶液等改进剂能够抑制解离,改善峰形。本文考察了0.01、0.025 和0.05mol/L 等不同浓度的磷酸二氢钾溶液,添加三乙胺调节p H 值为5.0、6.0 和7.0。结果磷酸二氢钾溶液浓度低,峰形拖尾较严重,磷酸二氢钾溶液浓度提高,色谱峰拖尾得到改善;p H 值越低,峰形越差,出峰时间越快;考察了乙腈的比例对保留时间的影响,乙腈的量15%的时候,保留时间约为40min;乙腈比例为20%时,保留时间约为16min,比较合适。
3)对左旋咪唑的稳定性进行了研究,取左旋咪唑对照品溶液(浓度 为500µg/mL)1.00mL, 分 别加1mol/L 盐 酸 溶 液、1mol/L 氢氧化钠溶液和5%过氧化氢溶液各1mL,放置12h,依法测定。结果左旋咪唑在这三种溶液中均稳定,几乎没有降解。
4)对含量较低或者存在非法添加物干扰的左旋咪唑注射液样品,非水滴定法多数无法准确测定其含量,而高效液相色谱法具有良好的专属性,能够准确测定左旋咪唑的含量,具有明显的优势。因此,建立的方法可用于左旋咪唑的质量控制。
5)高氯酸滴定液对温度敏感,需要根据温度做相应的校正,滴定体积较少,增加操作者的要求,滴定终点判断需要看颜色决定,有一定的误差;相对来说,高效液相色谱法操作简单、结果稳定、可靠,更适用于实践检验工作,值得推广。■

图4 左旋咪唑注射液供试品光谱图(批号20180901)

表1 HPLC 法与非水法测定含量结果比较(n=3)

表2 回收结果(n=5)
猜你喜欢
煤化工(2022年3期)2022-07-08
西部散文选刊(2022年1期)2022-02-03
合成纤维工业(2021年5期)2021-10-31
中华养生保健(2020年3期)2020-11-16
首都食品与医药(2020年1期)2020-10-21
农药科学与管理(2019年9期)2019-11-23
布达拉(2019年3期)2019-06-11
食品界(2017年4期)2017-05-17
山东工业技术(2016年10期)2016-09-06
中国质量万里行(2016年8期)2016-05-14
