
纳米碳材料催化乙苯脱氢制苯乙烯的研究进展
2018-04-12 01:04吴耿煌荣峻峰达志坚
石油学报(石油加工) 2018年2期
吴耿煌, 荣峻峰, 达志坚
(中国石化 石油化工科学研究院, 北京 100083)
苯乙烯是现代石油化工产品中最重要的单体之一。苯乙烯分子上的乙烯基化学性质活泼,容易发生加聚反应(包括自聚与共聚),其在工业上被广泛应用于生产聚苯乙烯、丁苯橡胶、丙烯-丁二烯-苯乙烯树脂、苯乙烯-顺丁烯二酸酐共聚物和不饱和聚酯等聚合物材料[1]。近年来,全球范围内以苯乙烯为原料制备的新型高分子化工产品的不断涌现,进一步推动了苯乙烯市场需求的增长,2015年全球苯乙烯的产能达到了33078 kt/a[2]。目前,工业上生产苯乙烯的主要方法为采用以含K2O助剂的Fe2O3为催化剂的乙苯催化脱氢法,该方法无论是催化剂、反应器还是工艺条件都十分成熟。但由于在生产过程中需要消耗大量的水蒸气(水蒸气与乙苯的摩尔比为8~10),这使得该工艺能耗过高[3]。在当前全球能源短缺、环境污染问题日益突出的形势下,如何实现苯乙烯生产过程中的节能降耗无疑具有十分重要的现实意义。
早在20世纪70年代,Iwasawa等[4]发现焦炭能够促进乙苯氧化脱氢反应。Cadus等[5]提出焦炭上的氧物种为碳材料催化乙苯氧化脱氢的活性中心。Grunewald等[6]将热解聚合物制备的碳催化剂应用于乙苯的氧化脱氢,乙苯转化率达到80%以上,苯乙烯选择性达到90%以上。尽管这些碳材料在反应初期都展现了较高的催化活性,但无定型的碳催化剂稳定性差且易积炭,活性下降很快,仅仅数小时后就会损失大部分的活性。2001年,Mestl等[7]首次以纳米碳纤维作为催化剂应用于乙苯的氧化脱氢反应。在他们的研究中,纳米碳纤维的催化活性高于高分散石墨,稳定性远远优于无定形的传统炭黑。至此,纳米碳材料作为一类新兴的绿色催化材料,在各种类型脱氢反应中的应用研究全面展开[8]。
纳米碳催化是直接使用纳米碳材料作为催化剂,并不负载或添加任何金属,活性中心为碳材料表面的缺陷结构与(或)官能团。相比于传统的无定形炭或活性炭,纳米碳材料表现出机械强度高,热稳定性好,导电和导热能力强,化学结构和酸碱性易于调控等特点。而与金属催化剂相比较,纳米碳材料作为催化剂还具有原料来源广泛、无重金属污染、环境友好等优点[9-10]。鉴于乙苯脱氢反应在工业上的重要意义以及纳米碳材料在该反应体系中具有十分广阔的应用前景,在本文中,笔者简要介绍了纳米碳材料的结构和表面化学性质,重点介绍了近年来纳米碳材料在催化乙苯的直接脱氢、氧化脱氢的研究进展,并对该领域存在的问题及前景进行展望。
1 纳米碳材料的结构和表面化学性质
纳米碳材料是指分散相尺度至少有一维处于纳米量级(1~100 nm)的碳材料,主要存在形式包括零维的富勒烯(Fullerene)、纳米金刚石(Nano diamond,ND),一维的碳纳米管(Carbon nanotubes, CNT)、碳纳米锥(Carbon nano cone)以及二维的(氧化)石墨烯(Graphene)等,图1为典型纳米碳材料的示意图[9]。

图1 典型纳米碳材料的示意图[9]Fig.1 Schematic illustration of some nanocarbon[9]
C的原子序数为6,其外层电子轨道结构为2s22p2。4个价电子在C相互结合或与其他原子形成化合物时,通过sp3、sp2、sp的杂化轨道,可以产生3种不同的化学键,即单键、双键和三键。不同的杂化轨道方式,赋予了不同碳材料各自特殊的性能。以石墨烯为例,C以六元环形式周期性排列于石墨烯平面内。每个C通过σ键与临近的3个C相连,s、px和py3个杂化轨道形成强的共价键合,组成sp2杂化结构,具有120°的键角,赋予石墨烯极强的力学性能。剩余的pz轨道在与平面垂直的方向彼此交叠,形成离域大π键。因此π电子可以在石墨烯晶体平面内自由移动,从而使得石墨烯具有良好的导电性[11]。图2是典型碳材料的电子轨道杂化方式相图[12]。
纳米碳材料通常通过电弧放电、化学剥离、化学气相沉积、高温裂解碳化、爆轰等剧烈过程而制得,其石墨或金刚石结构无法保持完整。制备的纳米碳材料经过化学方法纯化处理,缺陷和边界位置的C为了实现自身价键的饱和,容易与杂原子结合,形成特定的官能团修饰于碳材料的表面,进而使纳米碳材料具备一定的酸碱性质和氧化还原能力,极大地影响了纳米碳材料的物理、化学性能[13]。
由于在碳材料长期接触空气的过程中即可被O2缓慢氧化而形成含氧官能团,因而碳材料上的含氧官能团被广泛研究。此外N、B、S、P等元素也被科研工作者广泛应用于纳米碳材料的掺杂以调变纳米碳材料的性质。以石墨烯为例,图3为石墨烯表面可能存在的结构缺陷以及部分杂原子存在的化学形式示意图[14]。O与纳米碳材料缺陷位上的C结合,可以形成羟基、羧基、羰基、醛基、环氧基、桥氧基、酯基、酸酐等含氧官能团。这些官能团具有较高的化学活性,可作为纳米碳材料在多类催化反应中的活性中心。按亲电亲核性可将这些官能团分为两大类[15-17],其中亲核氧物种包括:羰基、酯基、内酯基和酸酐等中的C=O键;亲电氧物种包括:羟基、醚或桥氧基、羧基、醛基、内酯基和酸酐等中的C—O键。研究表明,亲核氧物种有利于吸附缺电子的烷烃、芳香烃反应物分子,并催化反应物氧化脱氢生成对应的烯烃分子,亲电氧物种则易吸附电子云密度高的烯烃产物分子,将烯烃深度氧化为CO和CO2等副产物,从而降低了反应的选择性[16]。
同O类似,B、N、S、P等非金属杂原子的掺杂会显著影响纳米碳材料的表面化学性质、电子云分布,周围化学环境等。这些杂原子掺杂改性后产生具有特殊性能的纳米碳材料已在电化学、催化、功能材料合成等领域得到了广泛应用[18-19]。N在纳米碳材料上主要以吡啶氮、吡咯氮、石墨氮和氨基氮等形式存在。由于N比C多出1个电子,掺杂N的碳材料变成富电子结构,而引入的含N官能团还可以增强碳材料的表面碱性。Chen等[20]将掺N的CNT应用于丙烷的氧化脱氢,发现N的掺杂可以提高催化剂活化O2的能力,降低总包反应能垒,显著地提高了催化剂的反应活性。与N类似,B的原子尺寸同样与C接近,只是B比C少了1个电子,因此B的掺杂使纳米碳材料具有部分缺电子特性。B在纳米碳材料中一般以单一的石墨结构形式存在,有助于提高纳米碳材料的石墨化程度和抗氧化能力。此外,相关研究还表明,B掺杂可以提高纳米碳材料催化丙烷氧化脱氢的选择性[21]。除了B、N以外,P掺杂的纳米碳材料在丁烷的氧化脱氢反应中同样较未掺杂P的纳米碳材料表现出更高的氧化脱氢选择性[16]。而已报道的S掺杂的纳米碳材料的应用主要集中在电催化领域,此类材料在烷烃、芳香烃的脱氢反应中的应用还有待更多的研究。值得注意的是,尽管纳米碳材料表面的缺陷位点反应活性高,但由于目前还无法有效地表征纳米碳材料表面缺陷位点的种类和数量,因此关于纳米碳材料缺陷位点对于催化性能的贡献仍存在许多争议[13]。

图3 石墨烯表面可能存在的结构缺陷以及部分杂原子存在的化学形式示意图[14]Fig.3 General model illustrating the possible structure of defects and doped heteroatoms on graphene[14]
2 纳米碳材料催化乙苯脱氢制苯乙烯
2.1 纳米碳材料催化乙苯氧化脱氢
乙苯的催化氧化脱氢,是指乙苯在催化剂和O2存在的条件下,生成苯乙烯和水,反应温度为400~550℃,其反应方程如下所示:
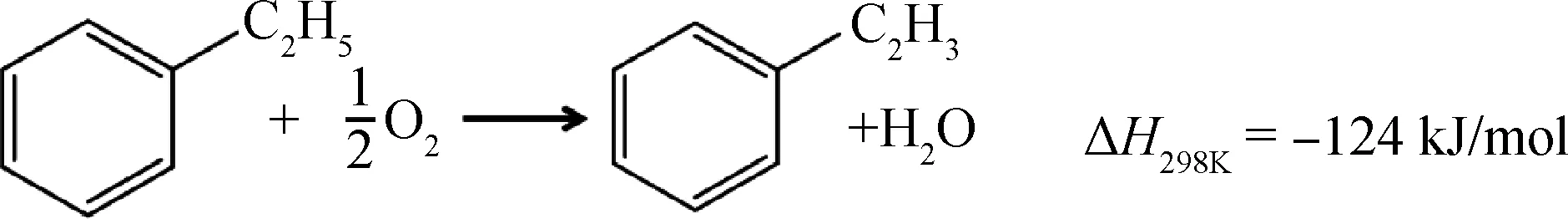
该反应为放热反应,不受热力学平衡限制。自1967年Distillers公司发表相关专利以来,科研人员已对乙苯的催化氧化脱氢反应进行了长期的探索[22]。早期的研究工作主要以金属磷酸盐和金属氧化物等材料作为催化剂的活性组分。2001年Mestl等[7]的研究表明,纳米碳纤维对乙苯的催化氧化脱氢具有优异的催化性能。Keller等[23]和Su等[24]又先后研究了具有sp2杂化的洋葱状纳米碳、CNT在催化乙苯氧化脱氢反应中的应用,2种纳米碳材料均体现较好的催化性能, 其活性接近甚至优于工业上的Fe2O3催化剂。2007年,Zhang等[25]进一步研究比较了sp2杂化的CNT和sp3杂化的ND以及活性炭催化乙苯氧化脱氢反应的性能,如图4[25]所示,ND表现出了类似于CNT的催化活性和稳定性,而活性炭则很快失活。以上研究表明,无论是sp2杂化或是sp3杂化的碳材料,只有长程有序结构的纳米碳材料才具有长期的催化活性和稳定性。此外,在上述的研究工作中还发现,纳米碳材料表面的含氧官能团与其催化性能有着密切的关联。

图4 纳米碳材料以及活性炭催化乙苯氧化脱氢反应速率-时间关系图[25] Fig.4 Time dependencies of ethylbenzene conversionrates of CNT, ND and activated carbon[25]
由于早期的纳米碳催化剂大多直接使用商业化的纳米碳材料,因此近年来的研究工作主要围绕着商业化纳米碳材料的掺杂改性以及新型结构纳米碳材料的制备及应用。Wang等[26]以经微波处理的玉米粒为前驱体,通过高温碳化的方式制备了一种蜂巢状结构微孔碳材料并将该材料应用于乙苯的氧化脱氢反应。该材料催化乙苯氧化脱氢的转化率为48%,产物苯乙烯的选择性为86%。Su等[27]则以介孔分子筛SBA-15为模板,蔗糖为C源,经过高温碳化后用HF移除SBA-15,制备了一种有序介孔碳材料。他们将这种材料应用于乙苯的催化氧化脱氢反应,乙苯转化率为69%,苯乙烯选择性为76%。他们发现,在催化乙苯氧化脱氢反应的诱导期,介孔碳表面会产生含有氧官能团的活性焦炭。通过一系列对比实验以及红外吸收光谱和X射线光电子能谱等表征数据的分析,他们认为,介孔碳上结构有序的碳单元及原位生成的焦炭对于材料的催化性能都起着重要作用,并且反应初期原位产生的含氧官能团是反应的活性位点。Qui等[28]发现,经过O3氧化处理过后的多壁CNT,其催化乙苯氧化脱氢的性能大幅提升,乙苯的转化率和苯乙烯的选择性分别达到了80%和92%,而原始CNT对应的转化率和选择性则仅有50%和68%。他们认为,O3处理过后的CNT表面有着更多的含氧官能团是提升催化性能的关键。Diao等[29]以多孔石墨烯为催化剂催化乙苯氧化脱氢,乙苯的转化率为65%,对应苯乙烯的选择性为93%。除了提及含氧官能团外,他们还认为多孔石墨烯上丰富的结构缺陷有利于催化剂活化O2并提升催化性能。此外,最近C与金属氧化物或其他非金属材料组成的复合催化材料的研究也取得了不错的进展。如Wang等[30]以乙醇为C源,通过化学气相沉积的方式在载体Al2O3表面沉积并包覆了具有sp2杂化结构的C层。这种C包覆Al2O3结构的催化剂催化乙苯氧化脱氢的转化率为65%,对应苯乙烯的选择性为95%。与CNT或ND等纳米碳材料相比,这种复合材料具有制备成本低、工艺简单等优点。Guo等[31]则以硼酸为B源、尿素为N源、葡萄糖为C源,在惰性气氛下高温碳化制备了一种C掺杂氮化硼(BN)片层纳米材料(BCN),其结构如图5[31]所示。相比于纯C材料,这种BCN复合材料具有十分优异的热稳定性,即使在800℃的空气气氛中,也无明显的质量损失。将其应用于乙苯的氧化脱氢,乙苯的转化率为54%,苯乙烯的选择性为89%。尽管BCN材料中C的质量分数不到20%,但相比于不含C的纯BN材料,其催化乙苯氧化脱氢的反应速率提升了一个数量级,说明了C的掺杂对于BCN的催化活性起着关键作用,同时BN单元则很好地提升了材料的稳定性。
图5 BCN材料理想结构示意图[31]Fig.5 The idealized structure of BCN[31]
2.2 纳米碳材料催化乙苯直接脱氢
乙苯的催化直接脱氢,是指乙苯在无O2条件下,由催化剂催化乙苯直接脱氢生成苯乙烯和H2。该反应是典型的吸热反应,受热力学平衡限制,其反应方程如下所示:
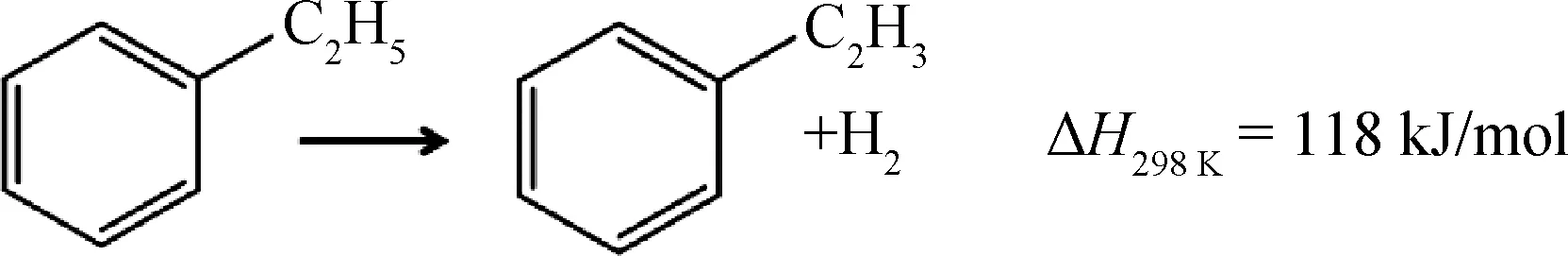
目前,工业上的乙苯脱氢催化剂的活性组分是含K2O助剂的Fe2O3催化剂,反应温度为600~700℃。为了维持Fe2O3活性相中Fe价态的稳定,同时消除积炭,在生产过程中需要通入大量的水蒸气(水蒸气与乙苯的摩尔比为8~10),产生大量的能耗[3]。相比于纳米碳材料在催化乙苯氧化脱氢中的研究历史,纳米碳材料催化乙苯直接脱氢的研究开展得较晚。2010年,Zhang等[32]首次将ND作为催化剂应用于乙苯直接脱氢制苯乙烯反应体系。他们发现,ND在550℃,无氧和无水蒸气保护的条件下,即可催化乙苯直接脱氢生成苯乙烯。如图6[32]所示,相同条件下,ND催化乙苯直接脱氢的反应活性为工业Fe2O3催化剂的3倍,对应的苯乙烯选择性则与之相当,达到了97.3%。更重要的是,反应过程中ND表面没有产生明显积炭且催化剂经过5次循环再生后其活性基本不变,这些结果表明了纳米碳材料在催化乙苯直接脱氢领域具有良好的应用前景。
Liu等[33]以沉积了CNT的碳化硅(SiC)材料为载体,负载ND。他们将这种复合型碳材料应用于乙苯的催化直接脱氢,获得了19.8%乙苯转化率和98.3%的苯乙烯选择性。而未负载的ND对应的乙苯转化率和苯乙烯选择性分别为15.1%和97.1%。他们认为复合材料具有良好的传热、传质性质,是催化剂性能提升的原因。同一时间,Thanh等[34]则是以氧化石墨烯为载体负载ND构筑了碳纳米复合材料。这种复合材料性能优异,催化乙苯直接脱氢的转化率为35.1%,对应的苯乙烯选择性为98.6%。随后Diao等[35]采用了类似的催化剂组成,并详细考察了ND负载量对催化剂性能的影响,他们发现,当ND的负载量为4%时,复合材料中每 1 g ND催化乙苯脱氢生成苯乙烯的反应速率达到了34.2 mmol/h。而ND负载量进一步增加到30%时,此数值则下降为9.4 mmol/h。该结果说明对于以ND为主要活性组分的复合型纳米碳催化剂,简单提高ND的负载量无法有效提高复合材料的催化性能。
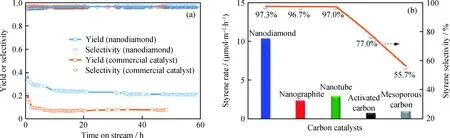
图6 纳米碳材料与工业Fe2O3催化剂、活性炭、介孔碳等材料催化乙苯脱氢反应性能比较图[32] Fig.6 Performance of catalytic dehydrogenation of ethylbenzene over carbon based catalysts and commercial Fe2O3(a) Performance of catalytic dehydrogenation of ethylbenzene over ND with commercial Fe2O3 catalyst for comparison;(b) Steady-state activities of various carbon catalysts[32]
除了不同纳米碳材料组成的复合材料外,针对ND、CNT等纳米碳材料的杂原子掺杂改性也是纳米碳材料催化乙苯直接脱氢反应近年来的研究热点之一[36-39]。其中,Zhao等[40-50]针对N掺杂、碳氮化合物(CNx)改性的纳米碳材料的制备及催化乙苯直接脱氢的应用研究开展了大量研究工作,于2014~2015年发表了系列研究论文。在CNx的制备方面,他们以SBA-15为模板,六亚甲基四胺为C、N源,通过惰性气氛下高温裂解碳化的方式,在SBA-15表面沉积N掺杂碳材料,之后用HF将SBA-15移除后,得到了有序介孔CNx[40]。而在纳米碳材料的N掺杂改性制备方面,他们先分别以三聚氰胺[41]、六亚甲基四胺[42]为N源,通过高温裂解的方式在ND表面沉积了CNx。随后他们选用了高温裂解过程更为剧烈的三聚氰胺硝酸盐[43]、三聚氰胺-三聚氰酸复合物[44]、以及六亚甲基四胺硝酸盐[45]为N源对CNT进行改性处理。这些N源前驱体除了对CNT进行N掺杂外,还通过剧烈的裂解反应,在CNT表面产生了大量的缺陷位点,进一步促进改性后的CNT的催化性能。此外,最近Liu等[51]以葡萄糖、碳酸铵、柠檬酸以及ND为原料,同样通过高温裂解的方式制备了一种ND@氮掺杂介孔碳复合材料,其制备过程如图7[51]所示。其中经过700℃高温裂解制备的复合材料催化乙苯直接脱氢的转化率37.7%,对应的苯乙烯选择性为99.6%。而900℃高温裂解制备的复合材料对应的乙苯转化率和苯乙烯选择性分别为31.7%和99.8%。这是目前已报道的纳米碳材料催化乙苯直接脱氢制苯乙烯所取得的最高选择性。他们认为ND在介孔碳中具有良好的分散性,有利于与反应物的充分接触,并且制备的复合材料表面的C=O官能团数量较为适中,是复合材料具有优异的催化性能的关键。
2.3 纳米碳材料催化乙苯脱氢的反应过程和机理
早在1988年,Cadus等[5]便提出了Al2O3在催化乙苯氧化脱氢过程中原位生成的焦炭为实际活性相,并给出了一种以碳材料表面氧官能团为活性位点的反应机理。然而由于早期相关表征手段的限制、焦炭自身稳定差等原因,这种机理更多的只是一种猜想。随后,Pereira等[52-54]以活性炭为催化剂催化乙苯氧化脱氢,并深入研究了催化剂活性位点、反应动力学参数以及催化剂失活现象。他们采用数值分析的方法对活性炭在He气氛下的程序升温脱附谱图进行分峰,并对碳材料表面的含氧官能团进行定量分析[52,55]。结果表明,活性炭催化乙苯氧化脱氢的反应速率与其表面所含羰基、醌基基团数量成正比。由此,他们认为,活性炭催化乙苯氧化脱氢的活性位点为碳材料表面的羰基、醌基基团。在2009年,Zhang等[56]的研究工作更为直接地证明了碳材料表面的醌基基团能够高效催化乙苯氧化脱氢。如图8[56]所示,他们以菲醌为前驱体,经过溴代、偶联2个步骤,制备了一种表面仅含醌式羰基基团的菲醌三聚体(Macrocyclic trimer,MCT)。MCT在350℃的反应温度下,单位表面积上催化乙苯脱氢的反应速率可达到金属氧化物、金属磷酸盐等的47倍,且高于工业上的Fe2O3催化剂。

图7 纳米金刚石@氮掺杂介孔碳(ND@NMC)复合材料制备示意图[51]Fig.7 Schematic illustration for the fabrication of the nitrogen-enriched mesoporouscarbon coated ND (ND@NMC) hybrid composite[51]
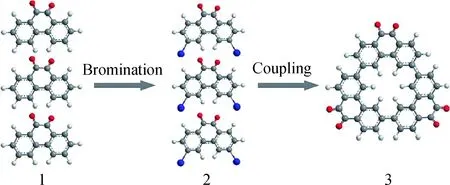
图8 菲醌三聚体的制备示意图[56]Fig.8 Schematic illustration for the fabrication ofmacrocyclic trimer of phenanthroquinone[56]
以上的研究结果证明,纳米碳材料表面的羰基基团在乙苯氧化脱氢反应中起着重要作用。然而,碳材料表面往往还含有一定量的羟基、羧基等含氧基团,上述研究无法明确这些官能团在碳材料催化乙苯氧化脱氢过程中所起的作用。为此,Qi等[57]结合有机化学和分析化学中对羰基、羟基和羧基的定量分析方法,开发出了一种半原位化学滴定方法,对氧化CNT(o-CNT)表面所含这3种含氧官能团的浓度进行定量分析。如图9[57](a)所示,他们以苯肼 (Phenylhydrazine, PH)、苯甲酸酐(Benzoic anhydride, BA)和2-溴苯乙酮(2-Bromoacetophenone, BrPE)为滴定剂分别选择性地与羰基、羟基和羧基进行反应。通过分析滴定剂的消耗量,可以推算o-CNT 表面这3种含氧官能团的浓度。图9(b)为经过滴定的o-CNT与原始o-CNT以及仅含缺陷的o-CNT(o-CNT(d))催化乙苯氧化脱氢性能的比较图。由图9可知,表面羟基与羧基的钝化对于o-CNT 的催化活性并无影响,而表面羰基钝化的o-CNT 的催化活性则显著下降。这一结果直接证明了羟基和羧基对o-CNT的催化乙苯氧化脱氢性能没有直接贡献,而羰基是o-CNT催化乙苯氧化脱氢的活性位点。之后他们又进一步开发了原位滴定的分析方法,并且证实了无论是CNT或者是石墨烯、洋葱状纳米碳等纳米碳材料,其表面的羰基基团都是催化乙苯氧化脱氢的位点,并且不同结构纳米碳材料表面的羰基基团的催化性能基本一致[58]。

图9 碳纳米管表面含氧官能团的化学滴定及对应的滴定衍生物催化乙苯氧化脱氢的性能比较图[57]Fig.9 Titration process for CNTS and the performance of ethylbenzene conversion of corresponding CNT derivatives[57](a)The titration processes for ketonic carbonyl, phenol and carboxylic acid groups on CNTs;(b) Ethylbenzene conversion rates of o-CNT, o-CNT derivatives, and CNT with only defects[57]
科研人员除了对纳米碳材料催化乙苯氧化脱氢的活性位点进行深入地研究外,还探究了纳米碳材料催化乙苯氧化脱氢的反应过程。Zhang等[25]以sp3杂化的ND和sp2杂化的CNT为催化剂催化乙苯氧化脱氢,考察了乙苯、O2分压以及反应温度对催化乙苯氧化脱氢的反应速率的影响。相关的动力学研究表明,单位比表面积上ND和CNT具有相近的催化乙苯氧化脱氢的反应速率、活化能及指前因子。这说明2种催化剂上活性位结构及其作用机理相同,且按照相同的反应路径催化乙苯氧化脱氢。此外,动力学模型的拟合表明,乙苯分子在CNT和ND上的催化氧化脱氢反应过程遵循双活性中心的Langmuir-Hinshelwood机理。该机理步骤包括:(1)乙苯和O2分别解离吸附在纳米碳材料表面2种不同的活性中心上(一般认为2种活性中心分别是羰基和石墨结构的缺陷位置),生成高活性的吸附态反应物;(2)吸附态的乙苯与O2在纳米碳材料表面发生氧化脱氢反应,生成吸附态产物苯乙烯和水;(3)吸附态的产物可逆脱附,完成了一个反应循环。他们进一步使用全氘代乙苯分子进行同位素示综动力学研究,发现步骤(2)中吸附态乙苯分子的乙基上的C—H活化断裂为整个反应的速控步骤。最近,Guo等[59]合成了一种新的模型化合物作为碳催化剂,结合原位滴定、O同位素示踪等分析方法,进一步研究了纳米碳材料的氧化脱氢过程。他们得出了一种新的结论:碳材料催化乙苯的氧化脱氢过程为(1)乙苯分子的乙基上C—H先在纳米碳材料表面羰基基团位上进行脱氢反应,同时羰基基团转变成羟基基团;(2)O2随后与脱下的H反应生成产物水,羰基活性位得以循环,且速控步骤仍为C—H的活化断裂步骤。通过以上研究可以看出,纳米碳材料表面的羰基基团为其催化乙苯氧化脱氢的活性位点,但纳米碳材料催化乙苯氧化脱氢反应的具体过程仍存在一定的争议,有待进一步深入的研究。
相较于纳米碳材料催化乙苯氧化脱氢的活性位点有着广泛的共识,目前关于纳米碳材料催化乙苯直接脱氢的活性位点则有缺陷和含氧官能团2种说法。Zhang等[32]在2010年以ND为催化剂催化乙苯直接脱氢时,提出了如下观点:ND表面的C在较大的表面曲率作用下发生部分石墨化,形成了独特的“金刚石-石墨烯”核壳纳米结构,且表层石墨烯的结构缺陷被大量的O饱和。反应过程如图10[60]所示,乙苯分子的乙基上C—H的H吸附在酮羰基氧上,再与羰基相互作用,C—H脱氢、羰基被还原为羟基,形成了一定数量的类取代芳香醇过渡中间体结构;此后产物苯乙烯分子发生脱附,而残留在催化剂表面的H则在高温条件下以H2形式脱附,实现了反应活性位的再生。除此以外,目前关于纳米碳材料催化乙苯直接脱氢的机理和过程的研究屈指可数。这一方面是由于相关表征手段的限制,另一方面则是由于纳米碳材料催化乙苯直接脱氢的温度比氧化脱氢温度高,碳材料在反应过程中存在一定的积炭现象且自身结构极易发生变化。尽管如此,相信随着相关研究的逐渐深入,今后关于纳米碳材料催化乙苯直接脱氢的反应过程及机理会有更加清晰、完整的认识。
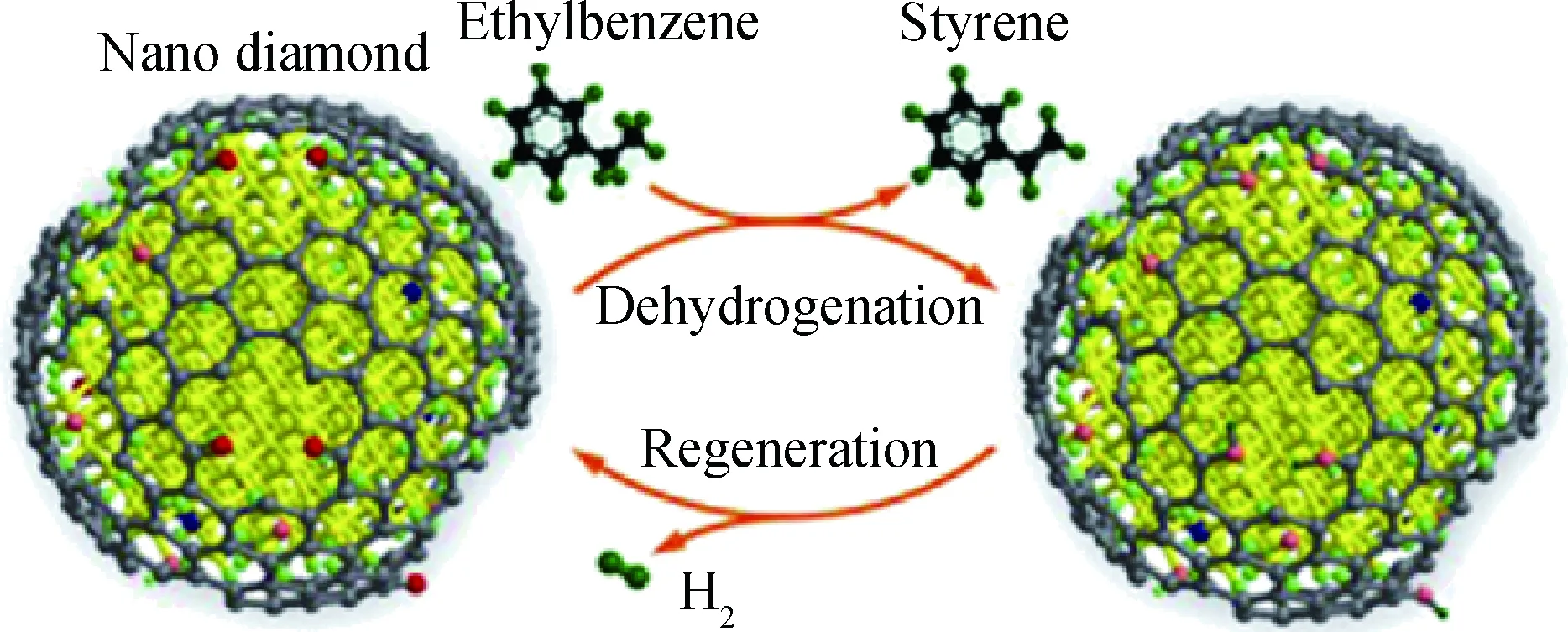
图10 纳米金刚石催化乙苯直接脱氢过程示意图[60]Fig.10 Schematic illustration for direct ethylbenzenedehydrogenation over ND[60]
3 结论与展望
乙苯脱氢制苯乙烯是工业上具有重要意义的脱氢反应。相比于传统的含K2O助剂的Fe2O3催化剂,纳米碳材料既可以在较低温度下高活性地催化乙苯氧化脱氢,也可以高选择性地催化乙苯直接脱氢,且在反应过程中均无需额外通入水蒸气,有效地提高了能源的利用效率。此外,纳米碳材料作为催化剂还具有结构调变能力强、原料来源广泛、无重金属污染、环境友好等优点。在能源日益匮乏的大背景下,发展纳米碳材料催化乙苯脱氢制苯乙烯无疑具有十分重要的研究价值,并且该领域的研究目前已取得了较大的进展,表现出了广阔的应用前景。
需要指出的是,进一步推动纳米碳材料催化乙苯脱氢制苯乙烯的工业规模应用之前,该领域仍存在许多挑战和关键性问题亟待解决:(1)以ND、CNT和石墨烯为代表的纳米碳材料制造成本依旧较高,并且大部分的研究将纳米碳材料直接作为纯相粉体使用,这直接导致纳米碳催化剂成本无法与当前工业化的Fe2O3催化剂竞争;(2)相关的基础研究表明,纳米碳材料上的含氧官能团以及缺陷是其催化乙苯脱氢制苯乙烯的活性位点。然而如何简单高效地规模化生产具有明确结构(包含定量的官能团及缺陷)的纳米碳材料仍没有理想的解决方案;(3)由于含氧官能团以及缺陷是纳米碳材料的催化活性位点,因此进一步提高这些位点的数量势必会导致纳米碳材料长程有序结构的破坏,导致催化剂的活性与稳定性存在着相互制约的难题。尽管近期的研究表明N、B等杂原子的掺杂有利于提高纳米碳材料的活性,但掺杂对于催化剂的稳定性、再生性的影响仍需进一步的研究。目前,纳米碳材料催化乙苯脱氢制苯乙烯虽然离规模化应用距离尚远,但这一新兴领域近年来发展迅速,各种新型(复合)碳材料不断涌现并且对相关反应认识日渐深入,相信纳米碳材料在这一领域的研究及应用将取得更多的进展与突破。
[1] 李皓巍, 金月昶, 金熙俊. 苯乙烯现状及工艺技术[J].当代化工, 2012, 41(9): 986-989. (LI Haowei, JIN Yuechang, JIN Xijun. Production and consumption status of styrene and its process technologies[J].Contemporary Chemical Industry, 2012, 41(9): 986-989.)
[2] 谭捷, 钟向宏. 国内外苯乙烯的供需现状及发展前景[J].石油化工技术与经济, 2016, 32(3): 13-17. (TAN Jie, ZHONG Xianghong. Supply and demand status of styrene at homeand abroad and its development prospect[J].Technology & Economics in Petrochemicals, 2016, 32(3): 13-17.)
[3] 刘剑锋, 缪长喜. 苯乙烯生产新技术研究进展[J].化学世界, 2013, 54(7): 442-447. (LIU Jianfeng, MIAO Changxi. Studies on energy saving technologies of styrene production[J].Chemical World, 2013, 54(7): 442-447.)
[4] IWASAWA Y, NOBE H, OGASAWARA S. Reaction mechanism for styrene synthesis over polynaphthoquinone[J].Journal of Catalysis, 1973, 31(3): 444-449.
[5] CADUS L E, ARRUA L A, GORRIZ O F, et al. Action of activated coke as a catalyst: Oxydehydrogenation of ethylbenzene to styrene[J].Industrial & Engineering Chemistry Research, 1988, 27(12): 2241-2246.
[6] GRUNEWALD G, DRAGO R. Oxidative dehydrogenation of ethylbenzene to stykene over carbon-based catalysts[J].Journal of Molecular Catalysis, 1990, 58(2): 227-233.
[7]MESTL G, MAKSIMOVA N I, KELLER N, et al. Carbon nanofilaments in heterogeneous catalysis: An industrial application for new carbon materials?[J].Angewandte Chemie International Edition, 2001, 40(11): 2066-2068.
[8] 苏党生. 纳米碳催化[M].北京: 科学出版社, 2014: 138-168.
[9] SU D S, PERATHONER S, CENTI G. Nanocarbons for the development of advanced catalysts[J].Chemical Reviews, 2013, 113(8): 5782-5816.
[10] YU D S, NAGELLI E, DU F, et al. Metal-free carbon nanomaterials become more active than metal catalysts and last longer[J].The Journal of Physical Chemistry Letters, 2010, 1(14): 2165-2173.
[11] RAO C, BISWAS K, SUBRAHMANYAM K, et al. Graphene, the new nanocarbon[J].Journal of Materials Chemistry, 2009, 19(17): 2457-2469.
[12] GEORGAKILAS V, PERMAN J A, TUCEK J, et al. Broad family of carbon nanoallotropes: Classification, chemistry, and applications of fullerenes, carbon dots, nanotubes, graphene, nanodiamonds, and combined superstructures[J].Chemical Reviews, 2015, 115(11): 4744-4822.
[13] SUN X Y, WANG R, SU D S. Research progress in metal-free carbon-based catalysts[J].Chinese Journal of Catalysis, 2013, 34(3): 508-523.
[14] HU H W, XIN J H, HU H, et al. Metal-free graphene-based catalyst-insight into the catalytic activity: A short review[J].Applied Catalysis A: General, 2015, 492(10): 1-9.
[15] TU N D K, CHOI J, PARK C R, et al. Remarkable conversion betweenn- andp- type reduced graphene oxide on varying the thermal annealing temperature[J].Chemistry of Materials, 2015, 27(21): 7362-7369.
[16] ZHANG J, LIU X, BLUME R, et al. Surface-modified carbon nanotubes catalyze oxidative dehydrogenation ofn-butane[J].Science, 2008, 322(5898): 73-77.
[17] 钟炳伟. 纳米碳材料在气相反应中的催化性能和机理研究[D].合肥: 中国科学技术大学, 2014.
[18] PARAKNOWITSCH J P, THOMAS A. Doping carbons beyond nitrogen: An overview of advanced heteroatom doped carbons with boron, sulphur and phosphorus for energy applications[J].Energy & Environmental Science, 2013, 6(10): 2839-2855.
[19] WANG H B, MAIYALAGAN T, WANG X. Review on recent progress in nitrogen-doped graphene: Synthesis, characterization, and its potential applications[J].ACS Catalysis, 2012, 2(5): 781-794.
[20] CHEN C L, ZHANG J, ZHANG B S, et al. Revealing the enhanced catalytic activity of nitrogen-doped carbon nanotubes for oxidative dehydrogenation of propane[J].Chemical Communications, 2013, 49(74): 8151-8153.
[21] FRANK B, ZHANG J, BLUME R, et al. Heteroatoms increase the selectivity in oxidative dehydrogenation reactions on nanocarbons[J].Angewandte Chemie International Edition, 2009, 48(37): 6913-6917.
[22] BRYAN W R, WILLIAM C C. Production of alkenyl benzenes: US, 3,318,966[P].1967-05-09.
[23] KELLER N, MAKSIMOVA N I, RODDATIS V V, et al. The catalytic use of onion-like carbon materials for styrene synthesis by oxidative dehydrogenation of ethylbenzene[J].Angewandte Chemie International Edition, 2002, 41(11): 1885-1888.
[24] SU D S, MAKSIMOVA N, DELGADO J J, et al. Nanocarbons in selective oxidative dehydrogenation reaction[J].Catalysis Today, 2005, 102(2): 110-114.
[25] ZHANG J, SU D S, ZHANG A H, et al. Nanocarbon as robust catalyst: Mechanistic insight into carbon-mediated catalysis[J].Angewandte Chemie International Edition, 2007, 46(38): 7319-7323.
[26] WANG L, ZHANG J, SU D S, et al. Simple preparation of honeycomb-like macrostructured and microporous carbons with high performance in oxidative dehydrogenation of ethylbenzene[J].Chemistry of Materials, 2007, 19(11): 2894-2897.
[27] SU D S, DELGADO J J, LIU X, et al. Highly ordered mesoporous carbon as catalyst for oxidative dehydrogenation of ethylbenzene to styrene[J].Chemistry-An Asian Journal, 2009, 4(7): 1108-1113.
[28] QUI N V, SCHOLZ P, KELLER T F, et al. Ozonated multiwalled carbon nanotubes as highly active and selective catalyst in the oxidative dehydrogenation of ethylbenzene to styrene[J].Chemical Engineering & Technology, 2013, 36(2): 300-306.
[29] DIAO J Y, LIU H Y, WANG J, et al. Porous graphene-based material as an efficient metal free catalyst for the oxidative dehydrogenation of ethylbenzene to styrene[J].Chemical Communications, 2015, 51(16): 3423-3425.
[30] WANG J, DIAO J Y, ZHANG J, et al. Few-layersp2carbon supported on Al2O3as hybrid structure for ethylbenzene oxidative dehydrogenation[J].Catalysis Today, 2018, 301: 32-37.
[31] GUO F S, YANG P J, PAN Z M, et al. Carbon doped BN nanosheets for the oxidative dehydrogenation of ethylbenzene[J].Angewandte Chemie International Edition, 2017, 56(28): 8231-8235.
[32] ZHANG J, SU D S, BLUME R, et al. Surface chemistry and catalytic reactivity of a nanodiamond in the steam-free dehydrogenation of ethylbenzene[J].Angewandte Chemie International Edition, 2010, 49(46): 8640-8644.
[33] LIU H Y, DIAO J Y, Wang Q, et al. A nanodiamond/CNT-SiC monolith as a novel metal free catalyst for ethylbenzene direct dehydrogenation to styrene[J].Chemical Communications, 2014, 50(58): 7810-7812.
[34] THANH T T, BA H, TRUONG-PHUOC L, et al. A few-layer graphene-graphene oxide composite containing nanodiamonds as metal-free catalysts[J].Journal of Materials Chemistry A, 2014, 2(29): 11349-11357.
[35] DIAO J Y, LIU H Y, FENG Z B, et al. Highly dispersed nanodiamonds supported on few-layer graphene as robust metal-free catalysts for ethylbenzene dehydrogenation reaction[J].Catalysis Science & Technology, 2015, 5(11): 4950-4953.
[36] WANG J, LIU H Y, DIAO J Y, et al. Size-controlled nitrogen-containing mesoporous carbon nanospheres by one-step aqueous self-assembly strategy[J].Journal of Materials Chemistry A, 2015, 3(5): 2305-2313.
[37] BA H, LIU Y F, TRUONG-PHUOC L, et al. N-doped food-grade-derived 3D mesoporous foams as metal-free systems for catalysis[J].ACS Catalysis, 2016, 6(3): 1408-1419.
[38] BA H, LUO J J, LIU Y F, et al. Macroscopically shaped monolith of nanodiamonds@ nitrogen-enriched mesoporous carbon decorated SiC as a superior metal-free catalyst for the styrene production[J].Applied Catalysis B: Environmental, 2017, 200: 343-350.
[39] SHI L, QI W, LIU W, et al. Carbon nitride modified nanocarbon materials as efficient non-metallic catalysts for alkane dehydrogenation[J].Catalysis Today, 2018, 301: 48-54.
[40] ZHAO Z K, DAI Y T, LIN J H, et al. Highly-ordered mesoporous carbon nitride with ultrahigh surface area and pore volume as a superior dehydrogenation catalyst[J].Chemistry of Materials, 2014, 26(10): 3151-3161.
[41] ZHAO Z, DAI Y T. Nanodiamond/carbon nitride hybrid nanoarchitecture as an efficient metal-free catalyst for oxidant-and steam-free dehydrogenation[J].Journal of Materials Chemistry A, 2014, 2(33): 13442-13451.
[42] ZHAO Z K, LI W Z, DAI Y T, et al. Carbon nitride encapsulated nanodiamond hybrid with improved catalytic performance for clean and energy-saving styrene production via direct dehydrogenation of ethylbenzene[J].ACS Sustainable Chemistry & Engineering, 2015, 3(12): 3355-3364.
[43] ZHAO Z K, DAI Y T, GE G F, et al. Facile simultaneous defect production and O, N-doping of carbon nanotubes with unexpected catalytic performance for clean and energy-saving production of styrene[J].Green Chemistry, 2015, 17(7): 3723-3727.
[44] ZHAO Z K, DAI Y T, GE G F, et al. Explosive decomposition of a melamine-cyanuric acid supramolecular assembly for fabricating defect-rich nitrogen-doped carbon nanotubes with significantly promoted catalysis[J].Chemistry-A European Journal, 2015, 21(22): 8004-8009.
[45] ZHAO Z K, DAI Y T, GE G F, et al. Increased active sites and their accessibility of a N-doped carbon nanotube carbocatalyst with remarkably enhanced catalytic performance in direct dehydrogenation of ethylbenzene[J].RSC Advances, 2015, 5(65): 53095-53099.
[46] ZHAO Z K, DAI Y T, GE G F, et al. A facile approach to fabricate an N-doped mesoporous graphene/nanodiamond hybrid nanocomposite with synergistically enhanced catalysis[J].Chem Cat Chem, 2015, 7(7): 1070-1077.
[47] ZHAO Z K, DAI Y T, GE G F, et al. Efficient tuning of microstructure and surface chemistry of nanocarbon catalysts for ethylbenzene direct dehydrogenation[J].AIChE Journal, 2015, 61(8): 2543-2561.
[48] ZHAO Z K, DAI Y T, GE G F, et al. Guanidine nitrate enhanced catalysis of nitrogen-doped carbon nanotubes for metal-free styrene production through direct dehydrogenation[J].Chem Cat Chem, 2015, 7(7): 1135-1144.
[49] ZHAO Z K, DAI Y T, GE G F, et al. Nitrogen-doped carbon nanotubes via a facile two-step approach as an efficient catalyst for the direct dehydrogenation of ethylbenzene[J].Physical Chemistry Chemical Physics, 2015, 17(29): 18895-18899.
[50] ZHAO Z K, DAI Y T, GE G F. Nitrogen-doped nanotubes-decorated activated carbon-based hybrid nanoarchitecture as a superior catalyst for direct dehydrogenation[J].Catalysis Science & Technology, 2015, 5(3): 1548-1557.
[51] LIU Y F, BA H, LUO J J, et al. Structure-performance relationship of nanodiamonds@ nitrogen-doped mesoporous carbon in the direct dehydrogenation of ethylbenzene[J].Catalysis Today, 2018, 301: 38-47.
[52] PEREIRA M, ORFAO J, FIGUEIREDO J. Oxidative dehydrogenation of ethylbenzene on activated carbon catalysts 1 Influence of surface chemical groups[J].Applied Catalysis A: General, 1999, 184(1): 153-160.
[53] PEREIRA M, ORFAO J, FIGUEIREDO J. Oxidative dehydrogenation of ethylbenzene on activated carbon catalysts 2 Kinetic modelling[J].Applied Catalysis A: General, 2000, 196(1): 43-54.
[54] PEREIRA M, ORFAO J, FIGUEIREDO J. Oxidative dehydrogenation of ethylbenzene on activated carbon catalysts 3 Catalyst deactivation[J].Applied Catalysis A: General, 2001, 218(1): 307-318.
[55] FIGUEIREDO J, PEREIRA M, FREITAS M, et al. Modification of the surface chemistry of activated carbons[J].Carbon, 1999, 37(9): 1379-1389.
[56] ZHANG J, WANG X, SU Q, et al. Metal-free phenanthrenequinone cyclotrimer as an effective heterogeneous catalyst[J].Journal of the American Chemical Society, 2009, 131(32): 11296-11297.
[57] QI W, LIU W, ZHANG B S, et al. Oxidative dehydrogenation on nanocarbon: Identification and quantification of active sites by chemical titration[J].Angewandte Chemie International Edition, 2013, 52(52): 14224-14228.
[58] QI W, LIU W, GUO X L, et al. Oxidative dehydrogenation on nanocarbon: Intrinsic catalytic activity and structure-function relationships[J].Angewandte Chemie International Edition, 2015, 54(46): 13682-13685.
[59] GUO X L, QI W, LIU W, et al. Oxidative dehydrogenation on nanocarbon: Revealing the catalytic mechanism using model catalysts[J].ACS Catalysis, 2017, 7(2): 1424-1427.
[60] 张建, 王锐, 苏党生. 纳米金刚石非金属催化性能研究进展[J].中国科学化学, 2012, 42(4): 406-414. (ZHANG Jian, WANG Rui, SU Dangsheng. Research progress on metal-free catalysis by nanodiamond[J].Scientia Sinica Chimica, 2012, 42(4): 406-414.)
猜你喜欢
中学生数理化·高一版(2022年4期)2022-05-09
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
沈阳化工大学学报(2020年4期)2020-04-06
云南化工(2019年2期)2019-05-16
石油炼制与化工(2018年12期)2018-03-21
当代化工研究(2016年5期)2016-03-20
化工进展(2015年6期)2015-11-13
中国塑料(2015年5期)2015-10-14
高中生学习·高二版(2015年3期)2015-05-21
世界热带农业信息(2014年11期)2015-01-05
