
人乳头瘤病毒E6蛋白通过上调氯离子蛋白通道5的表达促进宫颈癌细胞迁移
2018-04-03 08:49赵凯旋徐静云卢春
江苏大学学报(医学版) 2018年2期
赵凯旋,徐静云,卢春
(南京医科大学病原微生物学系,江苏南京211166)
高危型人乳头瘤病毒(human papillomavirus,HPV)持续感染是宫颈癌致病的关键因素,约99.7%的宫颈鳞状细胞癌由HPV感染引起[1-2]。高危型HPV感染宫颈上皮细胞后可编码两种重要的致瘤蛋白——E6和 E7蛋白,以维持细胞的转化特性[3-6]。研究显示,E6通过结合并泛素化降解肿瘤抑制基因p53,阻断细胞凋亡,E7则通过竞争性结合视网膜母细胞瘤蛋白pRb,促进转录因子E2F的释放,从而调控细胞周期[7-8],最终导致肿瘤的发生。随着 HPV E6蛋白在诱导宫颈癌发生的分子机制方面的研究不断深入,近年来的研究发现,HPV E6还可通过激活端粒酶活性、结合并降解含有PDZ结构域的蛋白等途径促进宫颈癌的发生发展[9]。深入研究HPV E6蛋白促进宫颈癌发生发展的分子机制将有助于发现诊断和治疗宫颈癌的有效靶标。
氯离子蛋白通道5(chloride voltage-gated channel 5,CLCN5)基因编码的蛋白主要定位于内体膜,可促进肾近端小管对白蛋白的摄取[10-12]。已有文献证明CLCN5基因的突变可导致Dent病[13]。然而,CLCN5基因在肿瘤尤其是在宫颈癌中的作用却未见报道。
本研究在 HPV18阳性的HeLa细胞中敲低HPV E6后检测CLCN5基因的表达。构建含CLCN5基因的重组质粒,并将重组质粒以及CLCN5的小干扰RNA(siCLCN5)分别转染宫颈癌HeLa细胞,观察CLCN5在宫颈癌细胞迁移过程中的作用。
1 材料与方法
1.1 细胞与试剂
人宫颈癌HeLa细胞和人胚肾293T细胞(中国科学院细胞库);pCDNA3.1-3×Flag(简称 pCDNA3.1)表达载体为本实验室保存;E6小干扰RNA(siE6)和siCLCN5由上海吉玛公司合成;实时荧光定量PCR(RT-qPCR)酶以及PCR所用的2×Phata HSMaster Mix购自南京诺维赞科技有限公司;PCR引物由南京铂尚生物技术有限公司合成;限制性内切酶Bam HⅠ和NotⅠ(日本TaKaRa公司);T4连接酶(美国Thermo Scientific公司);胎牛血清(Gibco公司);LipofectamineTM2000(Invitrogen公司);抗CLCN5单克隆抗体(南京巴傲得生物科技有限公司);抗α微管蛋白单克隆抗体(美国Santa Cruz公司);抗HPV18 E6抗体(Abcam公司);辣根过氧化物酶标记的羊抗兔IgG购自碧云天生物技术公司;Transwell小室(Millipore公司)。
1.2 E6和 CLCN5 siRNAs的转染
将E6 siRNA和CLCN5 siRNA冻干粉瞬时离心至管底,加入无RNA酶水配制成终浓度为20μmol/L的样品。选取生长良好的HeLa细胞接种于6孔板。次日,当细胞生长至70%汇合度时,用脂质体转染试剂LipofectamineTM2000分别将siRNAs及其阴性对照(NC)转染至HeLa细胞中,6 h后将细胞培养基更换为含10%胎牛血清的完全培养基。48 h后提取细胞总RNA和总蛋白,分别用于后续实验检测mRNA和目的蛋白的表达。
1.3 RT-qPCR检测E6和CLCN5 mRNA转录水平
用Trizol试剂分别提取转染阴性对照和E6 siRNA两组细胞总RNA,按照反转录试剂盒说明书将RNA反转录为cDNA。以反转录后的cDNA为模板,RT-qPCR检测E6和CLCN5mRNA的表达水平。反应体系:SYBR混合物10μL,上下游引物各0.4 μL,蒸馏水8.2μL。反应条件:95℃预变性30 s,95℃10 s、60℃30 s,共进行40个循环,最后以95℃15 s,60℃60 s,95℃15 s采集熔解曲线。
1.4 蛋白质印迹法检测HeLa细胞中E6和CLCN5蛋白的表达
收集转染阴性对照和E6 siRNA的细胞,提取总蛋白,进行聚丙烯酰胺凝胶电泳并转膜,经5%脱脂牛奶封闭后将PVDF膜浸在E6和CLCN5一抗(用一抗稀释液以1∶1 000的比例稀释)中,4℃孵育过夜。TBST洗膜后,用相应的二抗37℃孵育1 h,进行蛋白显色。
1.5 PCR扩增CLCN5基因
根据CLCN5基因的核酸序列设计PCR上下游引物。上游引物(下划线为 Bam HⅠ识别序列),下游引物A-3′(下划线为NotⅠ识别序列)。以293T细胞基因组DNA为模板,PCR扩增CLCN5序列。通过1%琼脂糖核酸电泳鉴定PCR产物并进行切胶回收。
1.6 过表达质粒pCDNA3.1-CLCN5的构建与鉴定
用限制性内切酶Bam HⅠ和NotⅠ分别双酶切载体pCDNA3.1和切胶回收的PCR产物,双酶切产物纯化后经T4DNA连接酶连接,连接产物转化入感受态大肠埃希菌DH5α中,涂板并挑取有氨苄西林抗性的菌落进行扩增,通过质粒小提试剂盒提取重组质粒,进行双酶切鉴定及核酸序列测定。
1.7 CLCN5在宫颈癌HeLa细胞中的表达
用脂质体转染试剂LipofectamineTM2000分别将空载体 pCDNA3.1和上述构建好的过表达质粒pCDNA3.1-CLCN5转染到宫颈癌HeLa细胞中,48 h后提取两组细胞的总蛋白,蛋白质印迹法验证标签蛋白和CLCN5蛋白的表达。
1.8 Transwell迁移实验
将Transwell小室置于24孔板中,向下室加入500μL含10%胎牛血清的DMEM培养基。将1×104个细胞重悬于200μL纯DMEM培养基中,并接种至Transwell上室中,置于37℃、5%CO2培养箱培养12 h后,甲醇固定15 min,结晶紫染色15 min,流水漂洗晾干后置于显微镜下拍照并计数细胞。
1.9 统计学分析
实验数据采用SPSS 19.0软件进行统计学分析,两组均数间的比较采用t检验,P<0.05为差异有统计学意义。
2 结果
2.1 敲低HPV E6对宫颈癌细胞中CLCN5表达水平的影响
RT-qPCR和蛋白质印迹检测结果显示,E6的mRNA和蛋白水平被明显敲低,且与对照组相比,敲低E6后CLCN5的mRNA和蛋白水平也显著下降(图1)。
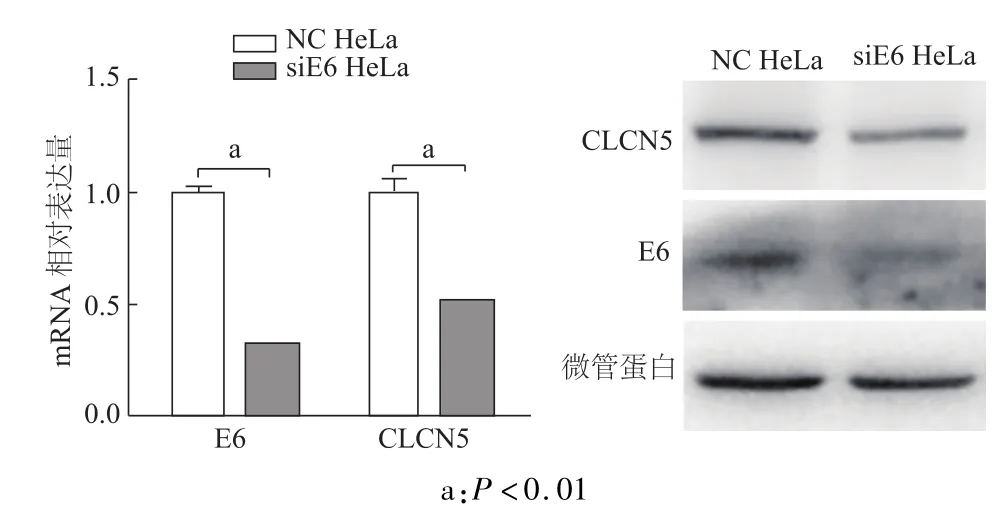
图1 敲低HPV E6对宫颈癌细胞CLCN5表达的影响
2.2 过表达质粒pCDN3.1-CLCN5的构建与鉴定
以293T细胞基因组DNA为模板,PCR扩增CLCN5基因,利用1%琼脂糖凝胶电泳对PCR产物进行鉴定,大小约2 490 bp(图2左),与预期条带大小一致。采用限制性内切酶Bam HⅠ和NotⅠ双酶切重组质粒pCDNA3.1-CLCN5,结果显示出两条特异性片段,大片段为载体pCDNA3.1,大小为5 508 bp,小片段为基因 CLCN5,大小为2 490 bp(图2右)。核酸序列测定及比对结果显示,插入的CLCN5基因序列正确,提示重组质粒 pCDNA3.1-CLCN5构建成功。

图2 重组质粒pCDNA3.1-CLCN5的构建与鉴定
2.3 过表达CLCN5对HeLa细胞迁移的影响
蛋白质印迹结果显示,CLCN5在HeLa细胞中过表达成功(图3A)。Transwell迁移实验结果表明,过表达CLCN5组的宫颈癌细胞的迁移数量明显多于对照组(P<0.01,图3B),提示过表达 CLCN5可以促进宫颈癌细胞迁移。
2.4 敲低CLCN5基因对HeLa细胞迁移的影响
蛋白质印迹结果显示,转染siCLCN5后,HeLa细胞中CLCN5蛋白的表达水平得到有效抑制。Transwell迁移实验结果显示,与对照组比较,敲低CLCN5后HeLa细胞迁移数量明显减少,表明敲低CLCN5可显著抑制宫颈癌细胞的迁移。见图4。
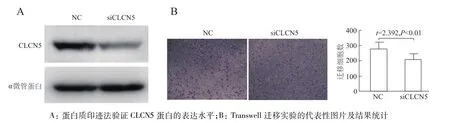
图4 敲低CLCN5对宫颈癌HeLa细胞迁移的影响
3 讨论
宫颈癌是导致女性癌症死亡的第3主要原因[14]。其致病因素多种多样,包括初次性生活的年龄、多个性伴侣、多次分娩、吸烟以及HPV的感染等。其中,高危型HPV的持续感染与宫颈癌的发生密切相关。HPV基因组包含早期基因区、晚期基因区以及长控制区,由早期基因区编码的E6、E7早期蛋白在病毒的生命周期及细胞转化中发挥了重要作用。
研究发现,HPV E6蛋白通过E6AP泛素途径降解p53是导致宫颈癌发生的重要分子机制[15-16]。HPV E6可通过调节钙黏蛋白转换促进宫颈癌细胞的迁移和侵袭[17]。且 E6可通过上调 miRNA-20b的表达,靶向抑制TIMP-2,从而增加宫颈癌细胞的侵袭力。E6还可活化钙调磷酸酶-NFAT信号促进宫颈癌细胞的增殖[18]。HPV E6靶向 TRIM25和USP15后可拮抗细胞质先天性免疫传感器RIG-1的活化[19]。同时,E6还可通过分泌促炎细胞因子和趋化因子修饰细胞微环境,从而影响免疫应答的功效[20]。充分了解和研究HPV E6蛋白的致病机制,对寻找预防和治疗宫颈癌的潜在靶标有重要意义。
既往研究结果表明,CLCN5基因突变可导致Dent病的发生,然而CLCN5基因在宫颈癌中的作用并未被阐明,且TCGA数据库的结果显示,CLCN5在宫颈癌中高表达。因此,我们猜测CLCN5可能参与调控了宫颈癌的发生、发展。本实验选取了HPV18阳性的HeLa细胞,采用RNA干扰技术敲低E6表达,结果显示,CLCN5 mRNA和蛋白水平均显著下调,提示HPV E6蛋白可能通过上调CLCN5的表达促进宫颈癌的发生、发展,过表达和敲低CLCN5后的功能学实验结果也初步证实CLCN5具有促进宫颈癌HeLa细胞迁移能力的作用。目前对于HPV E6蛋白调控CLCN5基因的分子机制有待于进一步研究,HPV E6蛋白可能通过结合并激活CLCN5启动子促进CLCN5的表达;HPV E6蛋白还可能通过促进 p53降解上调 CLCN5基因;此外,HPV E6蛋白可能通过抑制某些miRNAs的表达,使得CLCN5表达量增加,从而促进宫颈癌的迁移。
综上所述,本研究证实了 HPV E6蛋白对CLCN5基因的正调控作用,并且初步表明了CLCN5在宫颈癌细胞中具有促进细胞迁移的作用,为探索HPV E6蛋白调控CLCN5在肿瘤中的作用提供了线索,且CLCN5有望成为预防和治疗宫颈癌的新靶点。
[参考文献]
[1]Burd EM.Human papillomavirus and cervical cancer[J].Clin Microbiol Rev,2003,16(1):1-17.
[2]Goon PK,Stanley MA,Ebmeyer J,et al.HPV&head and neck cancer:a descriptive update[J].Head Neck Oncol,2009,1:36.
[3]zur Hausen H.Papillomaviruses in the causation of human cancers-a brief historical account[J].Virology,2009,384(2):260-265.
[4]Tilborghs S,Corthouts J,Verhoeven Y,et al.The role of nuclear factor-κB signaling in human cervical cancer[J].Crit Rev Oncol Hematol,2017,120:141-150.
[5]Zheng ZM,Tao M,Yamanegi K,et al.Splicing of a cap-proximal human papillomavirus 16 E6E7 intron promotes E7 expression,but can be restrained by distance of the intron from its RNA 5′cap[J].J Mol Biol,2004,337(5):1091-1108.
[6]Tang S,Tao M,McCoy JP Jr,et al.The E7 oncoprotein is translated from spliced E6*I transcripts in highrisk human papillomavirus type 16-or type 18-positive cervical cancer cell lines via translation reinitiation[J].JVirol,2006,89(9):4249-4263.
[7]Magaldi TG,Almstead LL,Bellone S,et al.Primary human cervical carcinoma cells require human papillomavirus E6 and E7 expression for ongoing proliferation[J].Virology,2012,422(1):114-124.
[8]McFarlane M,MacDonald AI,Stevenson A,et al.Human papillomavirus 16 oncoprotein expression is controlled by the cellular splicing factor SRSF2(SC35)[J].JVirol,2015,89(10):5276-5287.
[9]Ganti K,Broniarczyk J,ManoubiW,et al.The human papillomavirus E6 PDZ bindingmotif:from life cycle to malignancy[J].Viruses,2015,7(7):3530-3551.
[10]Picollo A,MalvezziM,Accardi A.Proton block of the CLC-5 Cl-/H+exchanger[J].JGen Physiol,2010,135(6):653-659.
[11]Satoh N,Suzuki M,Nakamura M,et al.Functional coupling of V-ATPase and CLC-5[J].World JNephrol,2017,6(1):14-20.
[12]Carr G,Simmons N,Sayer J.A role for CBS domain 2 in trafficking of chloride channel CLC-5[J].Biochem Biophys Res Commum,2003,310(2):600-605.
[13]Tang X,Brown MR,Cogal AG,et al.Functional and transport analyses of CLCN5 genetic changes identified in Dent disease patients[J].Physiol Rep,2016,4(8):e12776.
[14]Torre LA,Bray F,Siegel RL,et al.Global cancer statistics,2012[J].CA Cancer JClin,2012,65(2):87-108.
[15]Moody CA,Laimins LA.Human papillomavirus oncoproteins:pathways to transformation[J].Nat Rev Cancer,2010,10(8):550-560.
[16]McLaughlin-Drubin ME,Münger K.Oncogenic activities of human papillomaviruses[J].Virus Res,2009,143(2):195-208.
[17]Hu D,Zhou J,Wang F,et al.HPV-16 E6/E7 promotes cellmigration and invasion in cervical cancer via regulating cadherin switch in vitro and in vivo[J].Arch Gynecol Obste,2015,292(6):1345-1354.
[18]Ram BM,Dolpady J,Kulkarni R,et al.Human papillomavirus(HPV)oncoprotein E6 facilitates calcineurinnuclear factor for activated T cells2(NFAT2)signaling to promote cellular proliferation in cervical cell carcinoma[J].Exp Cell Res,2018,362(1):132-141.
[19]Chiang C,Pauli EK,Biryukov J,et al.The human papillomavirus E6 oncoprotein targets USP15 and TRIM25 to suppress RIG-I-mediated innate immune signaling[J].JVirol,2018,92(6):e01737.
[20]Iuliano M,Mangino G,Chiantore MV,et al.Human papillomavirus E6 and E7 oncoproteins affect the cell microenvironmentby classical secretion and extracellular vesicles delivery of inflammatory mediators[J].Cytokine,2017.Doi:10.1016/j.cyto.2017.11.003.
猜你喜欢
中老年保健(2021年12期)2021-08-24
江西农业学报(2021年4期)2021-04-20
水生生物学报(2021年1期)2021-02-04
中华养生保健(2020年10期)2021-01-18
中国生殖健康(2020年7期)2021-01-18
三农资讯半月报(2020年11期)2020-06-21
中国报道(2018年2期)2018-04-20
绿色中国(2016年1期)2016-06-05
中国当代医药(2015年9期)2015-03-01
中国中医药现代远程教育(2014年22期)2014-03-01
