
烟草种质的SSR标记遗传多样性及青枯病抗性的关联分析
2018-03-18 05:17:20赖瑞强李荣华夏岩石郭培国袁清华赵伟才
中国烟草学报 2018年6期
赖瑞强,李荣华,夏岩石,郭培国*,袁清华,赵伟才
1 广州大学生命科学学院 广州 510006;
2 广东省农科院作物研究所 广州 510640;
3 广东省烟草南雄科学研究所 广东南雄 512400
草青枯病是烟草生产中的一大毁灭性病害,主要发生在我国南方烟区。随着全球温室效应现象的加剧,高温高湿的情况越发严重,造成烟草发病率普遍提高,且该病害有向北蔓延扩展的趋势[1]。研究表明,烟草青枯病抗性为受多基因控制的数量性状,易受到基因型与环境交互作用的影响,使得培育青枯病抗病品种的任务更加严峻[2]。与传统青枯病抗病品种的选育方法相比,基于分子标记建立的辅助选择技术具有减少环境互作的影响、提高筛选效率等优点,可加速青枯病抗病品种的选育进程[2-3]。
基于家系群体开展连锁作图的QTL定位分析和基于连锁不平衡利用自然群体开展的关联分析是发掘与目标性状紧密连锁分子标记的主要方法。Nishi等[4]首次利用连锁分析发现1个与青枯病抗性相关的QTL位点,随后Qian等[2]、Lan等[5]和袁清华等[6]利用该方法分别探测到4、8和6个烟草青枯病抗性相关的QTLs。但由于连锁分析只限于检测来自双亲的2个等位变异,适用性较狭窄;而关联分析利用自然群体材料多世代的重组事件,能克服连锁分析存在的不足,具有较大的潜力发现复杂性状所牵涉到的多基因及相互关系[7]。使用关联分析方法,张吉顺等[8]和童治军等[9]成功发现了烟草一些农艺性状相关的等位基因,童治军等[9]、余义文等[10]、任民等[11]和樊文强等[12]在烟草一些化学成分研究中亦取得一些成绩;但烟草烟草青枯病抗性关联分析的研究较少,仅见吴超等[3]利用MFLP和SSR标记开展烟草青枯病抗性关联分析时发现到 6个MFLP标记位点与烟草青枯病抗性显著相关,但未发现有SSR标记位点与烟草青枯病抗性相关。众所周知,SSR标记具有多态性高、重复性好和呈共显性等优点[13-14];且其操作和检测也较为简单,只需通过PCR扩增和聚丙烯酰胺凝胶或琼脂糖凝胶电泳检测[15]。
群体结构分析是开展关联分析的基础。在烟草群体结构分析研究中,张吉顺等[16]利用16对SRAP引物将276份烟草种质划分为7个亚群,而童治军等[9]和樊文强等[12]利用SSR标记分别将96份和231份烟草种质划分为3个亚群;然而基因组内存在多个分子标记紧密连锁的不平衡区域[17],将这些区域当做一个遗传标记(即一个单倍型)进行研究,可减少研究的位点数[17-18];而且通过构建单倍型,可将存在强连锁不平衡的几个位点当作一个超位点进行群体结构分析和关联分析,对性状的检测效能要比单标记高[19-20]。
据此,本研究利用SSR分子标记对94份烟草种质材料进行遗传多样性分析,获悉烟草种质材料间的遗传差异和进行单倍型的构建;在分析这些种质材料群体结构基础上,开展烟草青枯病抗性的关联分析;研究结果可为烟草种质的合理利用及青枯病抗病材料的筛选提供参考。
1 材料与方法
1.1 材料
94份烟草种质材料来源于美国、日本、加拿大、索马里、澳大利亚、津巴布韦以及中国等地,包括烤烟、晒烟和白肋烟3种类型,由广东省烟草南雄科学研究所提供,其详细信息见表1。
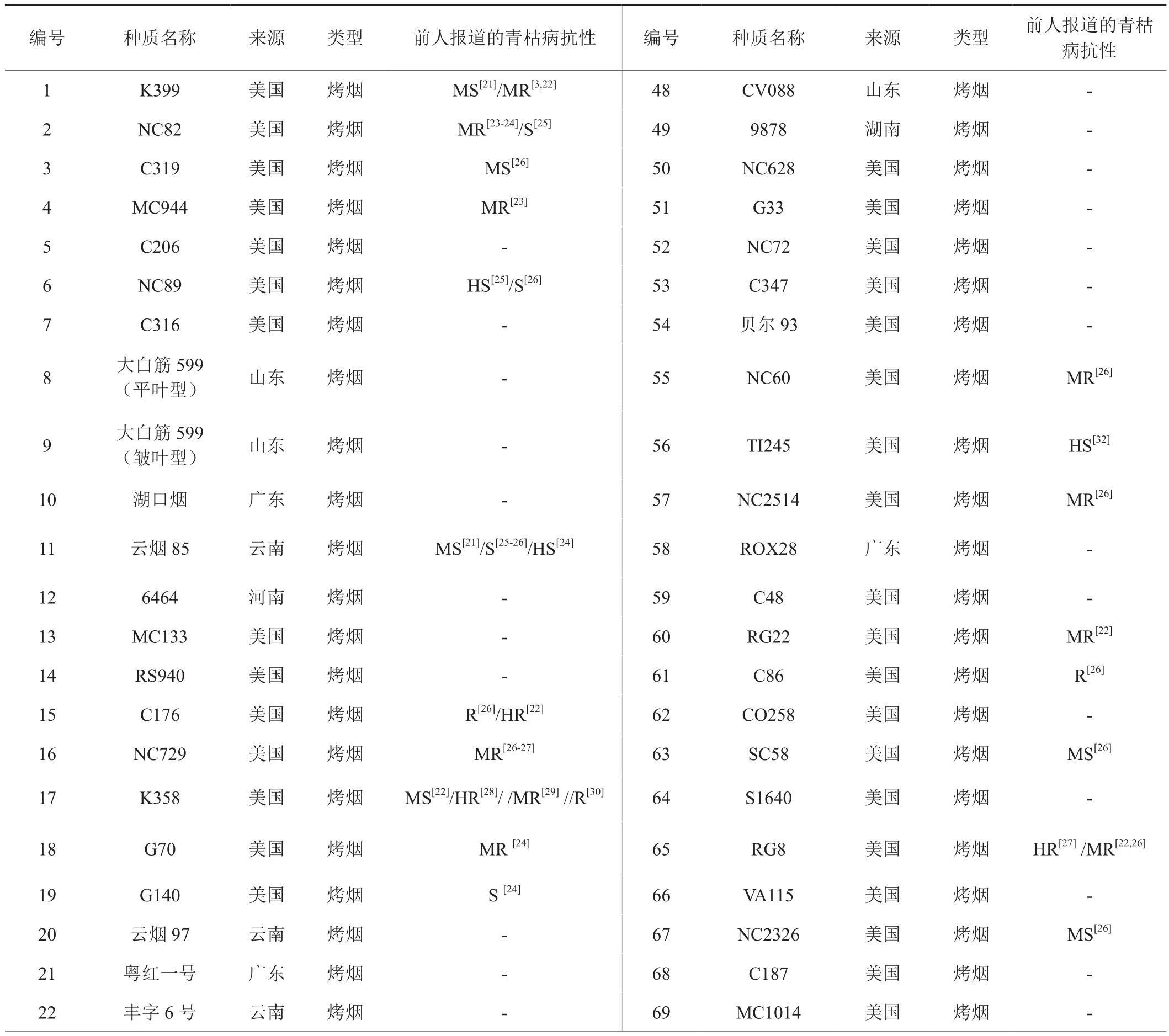
表1 94份烟草种质材料的来源与类型Tab.1 The origins and types of 94 tobacco germplasm materials

续表1
1.2 试验设计
在2013年3月9日和2014年2月29日移栽种植供试的94份烟草种质材料于广东省南雄烟草研究所试验田青枯病病圃,每份种质种植20株,采用随机区组设计,按常规方法栽培管理,以“岩烟97”和“红花大金元”分别作为抗病和感病对照品种。
1.3 青枯病病情调查
2013年5月8日和2014年5月4日使用茎部穿刺接种法[34]对处于旺长期的烟草进行人工注射基部茎杆接种致病型小种1、生化型III的青枯病病菌。2013年5月30日和2014年5月29日进行病情等级调查,调查标准参照《GB/T23222-2008烟草病虫害分级及调查方法》。并根据烟草病害等级计算病情指数(病指,Disease index,DI)[35]。
病指=∑(各级病株数×该病级值)/(调查总株数×最高级值)×100
据此,以《YC/T 41-1996烟草品种抗病性鉴定》的标准方法来划分94份种质的抗病性。
1.4 SSR分子标记技术
采取各种质材料的幼嫩叶片,使用改良的CTAB法[36]提取这些材料的DNA;采用琼脂糖凝胶电泳检测DNA质量、使用NanoDrop紫外分光光度计检测DNA浓度,并将所提取DNA稀释至20 ng·L-1,作为SSR扩增模板。
根据业已报道的烟草SSR引物等信息[37-41],从中筛选出在“岩烟97”、“红花大金元”、“118-3”和“粤烟98”四个材料中扩增条带清晰易辨、特异性良好且具有多态性的236对SSR引物(见网络版补充材料附表1)用于本研究。
SSR扩增的PCR反应体系为10 μL,包括DNA模板2μL,1μmol·L-1正向引物0.3μL,1 μmol·L-1反向引物0.3 μL,25 mmol · L-1MgCl20.8μL,Taq DNA Polymerase (5 U · μL-1)0.1 μL,10×PCR Buffer 1 μL,10 mmol · L-1dNTPs0.3 μL,双蒸水5.2μL。PCR扩增程序为94℃预变性 5 min,接着以94℃ 45s、相应引物退火温度45s和72℃ 1min,循环38次;最后72℃充分延伸10 min。采用6%非变性聚丙烯酰胺胶电泳和银染法[42-43]检测PCR扩增产物,所用试剂均购自于生工生物工程(上海)股份有限公司。
1.5 数据处理
使用BenQ M800扫描仪扫描凝胶电泳图谱,通过GelBuddy软件[44]判读凝胶电泳图谱中的条带,根据条带的有无,确定等位变异位点,并构建1、0二元数据矩阵。SSR变异位点的多态性信息量(Polymorphism Information Content,PIC) 的 计 算方法为PIC=1-∑Pa2,式中Pa表示出现第a个等位变异的频率[14]。利用Haploview软件[45],将SSR标记的等位变异位点1、0数据转换为“1”和“2”并制成数矩阵,选择该软件中的“Haps format”程序,并设置条件参数为位点间距小于500kb,缺失率低于50%,利用子程序“LD Plot”进行位点间的连锁不平衡分析,选择满足位点间连锁不平衡相关系数r2值大于0.8的强连锁区域,接着利用子程序“Haplotypes”构建单倍型。运用POPGENEVersion1.31软件计算烟草材料间的遗传一致度。运用Structure2.3软件对94份烟草种质进行群体结构分析,估计最佳的群体类群数K;设定K取值范围为1-11,观察随K值增加后验概率值LnP(D)的变化拐点来确定K值,如LnP(D)持续增加未出现拐点,则参照Evanno等[46]的方法以△K的最大值来确定K值并获取相应的群体矫正系数Q值。运用SPAGeDi1.3d软件[47]获取烟草种质间的亲缘系数,并将其中所有负值设为0,制成矩阵和Q值一起作为协变量进行群体矫正,再利用TASSEL3.0的混合线性模型(Mixed Linear Model,MLM)进行青枯病关联分析。
2 结果与分析
2.1 烟草青枯病病情统计及分析
2013年和2014年94份烟草自然群体青枯病病情调查结果见表2。从表中可见,94份材料两年间抗性评价一致的种质共59份,占总种质的62.77%,如“TI245”和“C206”、“C316” 等两年均表现出中抗(MR:25<DI≤50)或高抗(HR:0<DI≤25)青枯病,而“MC944”、“湖口烟”和“KE1”等两年均表现出中感(MS:50<DI≤75)或高感(HS∶75<DI≤100)青枯病;经pearson相关性分析,发现两年病指相关系数(PS)为0.691,达到极显著水平(P<0.01)。表明供试烟草种质两年间的抗病程度较为稳定。且2013年和2014年青枯病病指统计分析结果(表3)表明,病指变幅大,变异系数均较大,分别为89.69%和91.61%。表明94份烟草种质对青枯病的抗性变异较大,适合开展烟草青枯病抗性关联分析。
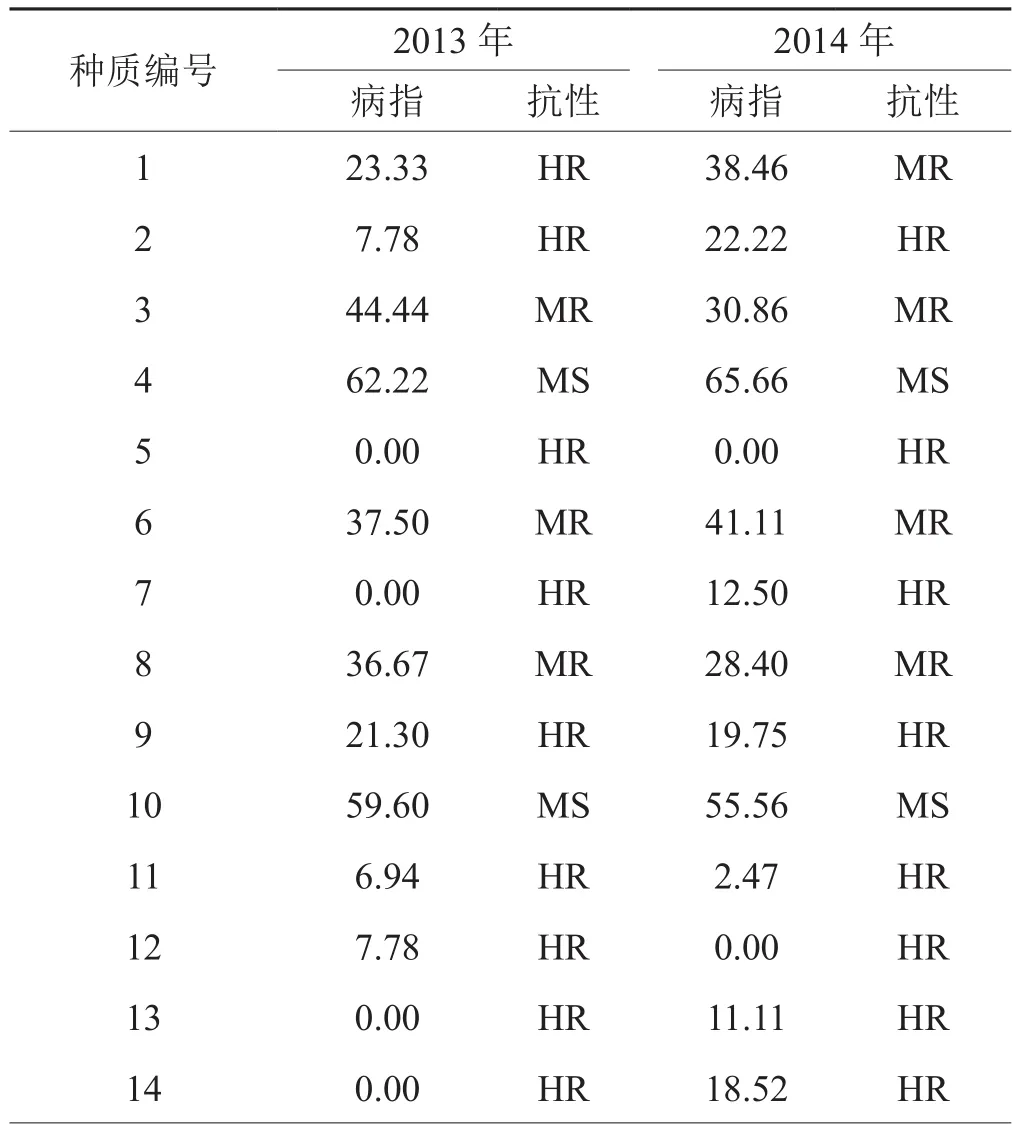
表2 供试烟草种质青枯病病情指数与抗性评价Tab.2 Bacterial wilt disease index of selected tobacco germplasm materials and resistance evaluation
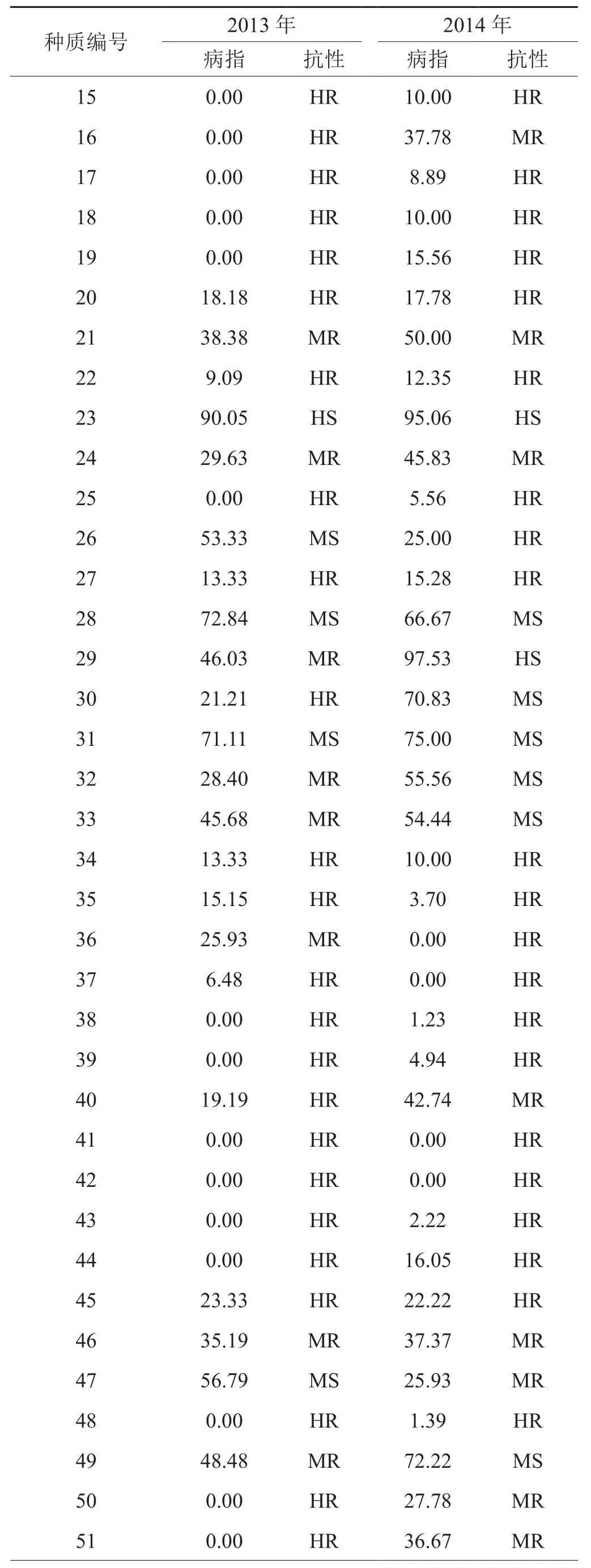
续表2
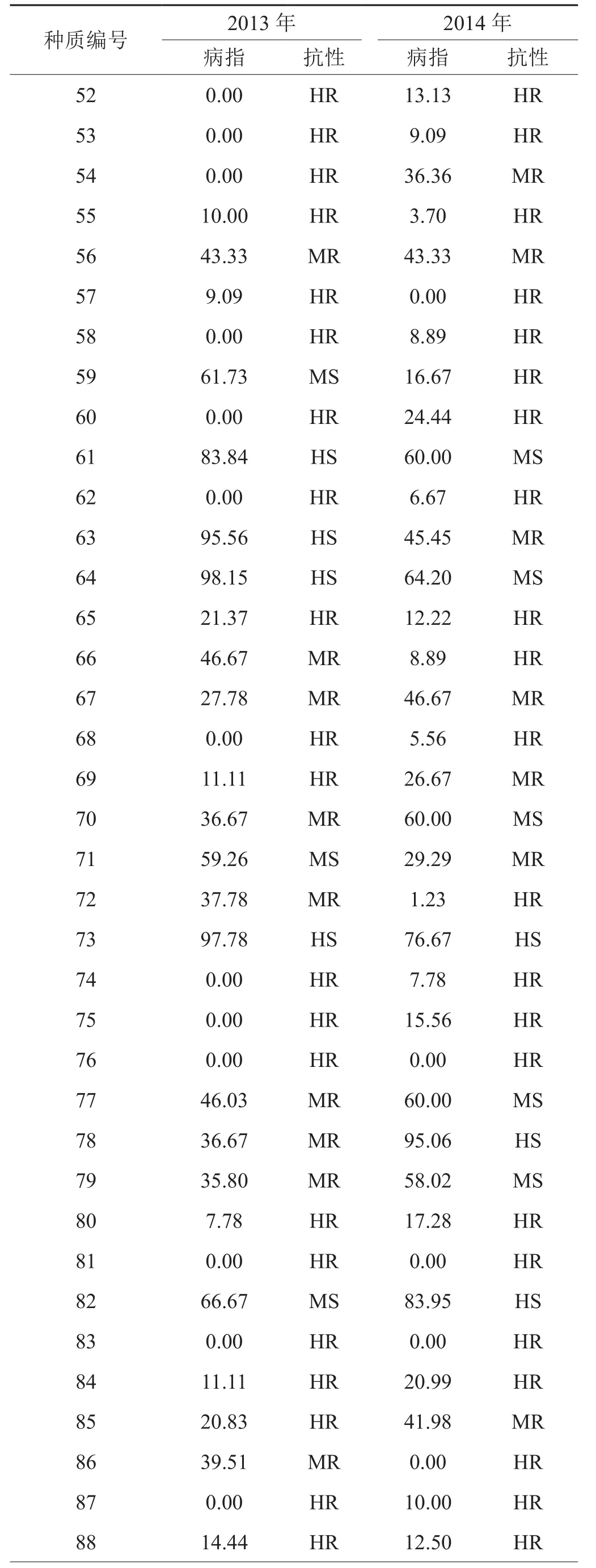
续表2
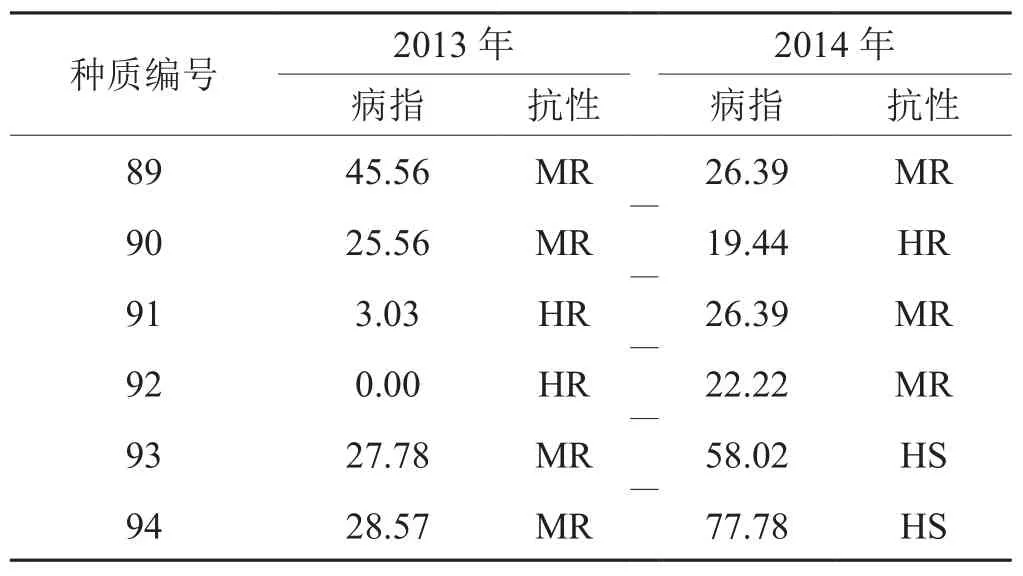
续表2

表3 供试烟草种质青枯病病情指数的统计分析Tab.3 Statistic data of bacterial wilt disease index of selected tobacco germplasm materials
2.2 SSR标记的多态性分析
使用聚丙烯酰胺凝胶电泳银染法检测各个SSR标记在94份种质材料中存在的等位位点(图1),236对SSR引物共检测到904个等位变异位点,各引物变异位点数目变化范围为1~8,平均变异位点数为3.83;数据分析表明SSR标记的PIC值变幅为0.1361~0.8760,平均为0.5136。表明这些引物具有较高的多态性信息,能在一定程度上反映烟草种质蕴藏的丰富基因信息。
2.3 基因多样性及亲缘关系分析
基因多样性是生物多样性的重要组成部分,对其研究有利于保护、开发和利用种质资源[48]。基于904个SSR标记位点,通过SPAGeDi1.3d软件计算可知94份烟草种质两两组合为4371个,其亲缘系数小于0.05的组合占总组合的72.41%,且亲缘系数为0的组合占了57.52%,表明大多数供试种质间亲缘关系弱或无亲缘关系。遗传一致度检测发现94份烟草材料的基因多样性指数为0.5405,即该群体随机抽取两个等位基因为杂合的概率均超过50%,表明所选种质的基因多样性较为丰富。

图1 SSR标记TM10788在94份烟草种质材料扩增产物的聚丙烯酰胺凝胶电泳银染图Fig.1 Silver staining image of polyacrylamide gel electorphoresis of SSR marker TM10788 in amplification products of 94 tobacco germplasm materials
2.4 单倍型的构建及群体结构分析
连锁不平衡分析中r2值是反映两个位点之间的相关系数,与关联分析的效力密切相关[49]。在本研究中,利用Phillips等[50]的连锁不平衡法对904个SSR位点进行单倍型的构建,选取r2值作为衡量标准,在r2大于0.8的强连锁不平衡条件下构建了61个单倍型块(D1~D61),这些单倍型块中含有126个单倍型(Hap1~126),且每个单倍型出现的频率均高于0.05。
基于126个单倍型,进行群体结构分析。设定类群数目K的取值范围为1~11,将MCMC(Markov Chain Monte Carlo)的不作数迭代(Length of burn in period)设为100000,对每个K值进行10次运算,获取后验概率值LnP(D),分析发现,随着K值的上升,后验概率LnP(D)表现出持续攀升的趋势而未出现明显的拐点(图2A),难以确定该群体的类群数;在此情况上,依照Evanno等[46]认为最大△K值时的K值即为最佳类群数的原则,分析发现,当△K值达到最大时对应的K值为2(图2B),表明该群体划分的最佳类群数为2,即类群Ⅰ和类群Ⅱ。

图2 K值与LnP(D)值(A)和△K值(B)的变化分布图Fig.2 Change distribution of K value with LnP(D) value (A) and△K value (B)
构建的群体结构图如图3所示。从中可见,类群Ⅰ由38份均为烤烟的烟草种质组成,这些种质来自美国26份,广东5份,云南3份,山东、福建、贵州和湖南各1份;类群Ⅱ包含56份烟草种质,由52份烤烟、2份广东晒烟和2份美国白肋烟组成,烤烟分别来源于美国24份,广东11份,山东5份,云南3份,河南2份,福建1份,津巴布韦2份,日本、索马里、加拿大和澳大利亚各1份。不同来源的材料在每个类群中均有分布,表明品种的类群划分结果和地理来源之间没有必然联系,与罗凯等[51]的研究结果一致。供试群体被分为两个类群,与前人[3,8,12,16]的研究相比,群体结构较简单,而相对简单的群体结构,有利于减少由于群体结构的影响而造成关联分析的假阳性,可提高关联分析的效果[52]。

图3 94份烟草种质的群体结构图Fig.3 Diagram of population structure of 94 tobacco germplasm materials
2.5 烟草青枯病抗性的关联分析
利用群体结构分析获得的各个体的矫正系数Q值和亲缘系数作为协变量进行校正,是避免基因型和表型在关联分析中出现假阳性的有效方式[8,47]。本研究中,根据基于126个单倍型分析所得的群体结构Q值和亲缘系数,利用Tassel软件对126个单倍型分别与2013年和2014年青枯病抗病性表型数据进行关联分析,结果发现2013年在P<0.05显著水平有4个单倍型与青枯病抗病性密切相关,对青枯病病指的表型变异解释率在8.19~13.65%之间(表4),其中Hap1、Hap64和Hap126这3个单倍型达到P<0.01的极显著水平,这3个单倍型对青枯病的表型变异解释率分别为13.65%、、13.56%和14.31%;2014年在P<0.05显著水平有7个单倍型与青枯病抗病性密切相关,对青枯病的表型变异解释率在 5.14~10.53%之间,其中有1个单倍型Hap62达到P<0.01的极显著水平,其对青枯病表型变异解释率为10.53%。这样,两年共检测到7个与青枯病抗病性密切相关的单倍型,其中两年均检测到的单倍型为三个,即Hap61、Hap62和Hap64,它们对对青枯病的表型变异解释率介于8.19~13.56%之间,平均达10.27%。
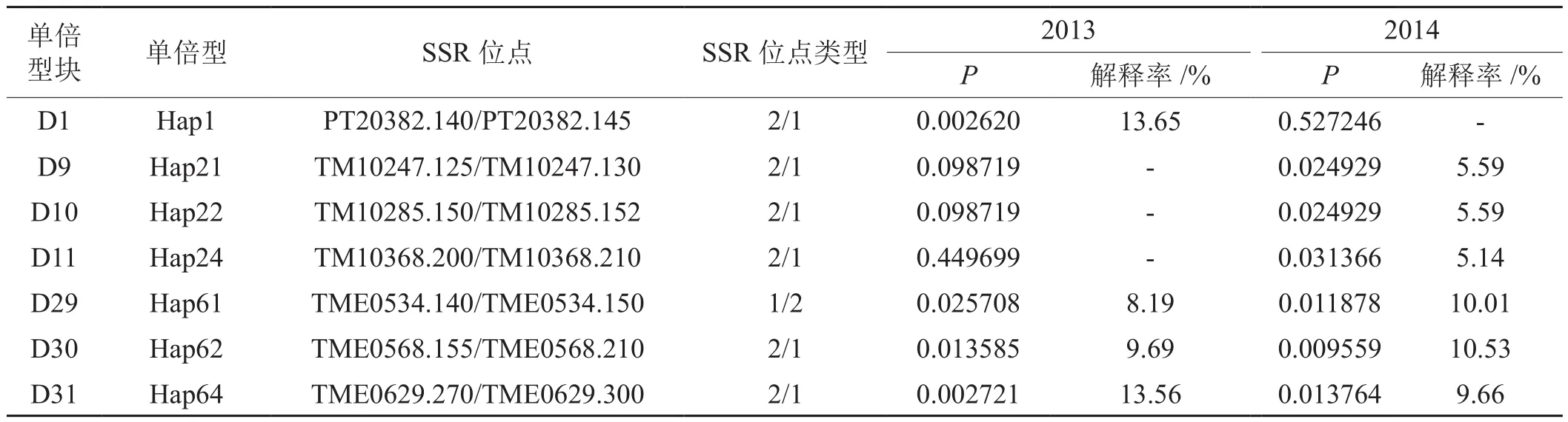
表4 与烟草青枯病抗性相关的单倍型及类型Tab.4 Haplotypes and loci associated with resistance of tobacco bacterial wilt
3 讨论
前人研究表明我国抗青枯病的优质烟草资源匮乏[53],且目前烟草青枯病抗源主要来自美国品种TI448A[54],抗源单一,遗传基础薄弱,不足以应对遗传多样性丰富且变异能力强的青枯病菌。因此通过研究烟草的遗传多样性,进而挖掘和利用其优良抗病基因至关重要。本研究对94份烟草种质进行遗传多样性分析,发现其基因多样性指数为0.5405,高于张吉顺等[8]、王国平等[55]和王一伊等[56]所选种质的基因多样性指数。表明供试的关联作图群体遗传多样性较丰富,符合Mackay等[57]提出进行关联分析时,需选取遗传多样性丰富的群体的理论。另外,从供试种质材料青枯病抗性来看,两年田间鉴定评价结果表明种质材料间的抗病性差异明显,多数与前人研究报道的抗病性基本一致,如感病材料“红花大金元”[21-27]和“许金1号”[22],抗病材料“岩烟97”[22,26-26,32]、“NC60”[26]和“930032”[28]等;部分种质与前人鉴定结果存在差异,如本研究鉴定“K326”为抗病材料,一些研究报道[3,23-24,28,31]与本研究所得结果一致,但另一些研究[21,25-27]表明其为感病材料;张振臣等[22]将“C86”鉴定为抗病材料,而本研究将其鉴定为感病材料等。导致鉴定评价结果差异的原因可能与接种的青枯病菌类型[25,33,58]、接种方式[59]、接种时烟草的生育期[59]和接种的菌液浓度[59-60]有关;此外,不同烟草病情等级划分及抗性鉴定标准[3,21-33]亦导致不同的评价结果。但本研究所有种质的鉴定标准具有统一性,种质间病指的差异主要来源于自身的抗性能力,且两年间的病指存在显著相关,烟草种质抗性鉴定一致性比例大于60%,表明本研究病指可靠,抗性鉴定结果具有一定的参考价值,利用该病情指数进行关联分析可靠性高。
地理环境的差异和自然选择压力的存在导致不同来源的材料间存在一定的群体结构,继而造成群体内出现多个类群的情况。本研究对94份烟草种质进行群体结构分析,将该群体划分为2个类群,群体结构较简单,每个类群均包含不同地理来源的烟草种质,表明不同地理来源的种质在群体结构上的分化不明显,与前人[51]的研究结果一致;发现来源地相同的烟草材料并不能完全聚在一起,部分美国烤烟和中国的烤烟被归在同一类群,而中国不同省份的烤烟在相同的类群中均有分布,晒烟和白肋烟被归为一类,该研究结果符合吴超等[3]和刘文杰等[61]的研究结果。说明本研究的群体结构分析结果可靠,由此获得的矫正系数Q值可信度高,可以用于后续的关联分析,以减少群体结构对关联分析准确度的干扰。
关联分析利用自然群体材料多世代的重组事件,有利于发现复杂性状与分子标记间的关系。然而烟草青枯病受微效多基因控制,大多数单标记的作用往往很小,难以通过严格的检验标准,且染色体上的等位基因间存在不完全的连锁不平衡,导致利用单标记进行关联分析时,仅有少数单标记与性状显著关联[62]。但基于单倍型的关联分析,其单倍型由多个单标记组成,区组信息和互作信息利用较为充分,能够克服单标记关联分析的局限性,具有更好的检测效能[63-64]。然而以往研究多采用单标记进行相关性状的关联分析,如童治军等[9]基于SSR单标记对231份烤烟的8个农艺性状和5个化学成分进行关联分析,仅发现到9个重要的QTL位点。而如果利用单倍型关联分析,可能会检测到更多与目的性状显著关联的QTL位点;如任民等[11]基于62个SSR位点对烟草致香物质进行单标记和单倍型的关联分析,发现在单标记关联分析中未被关联到的3个单标记,在其构建为单倍型后被显著关联,且关联到的致香成分最多。
由于关联分析是以连锁不平衡为基础的QTL定位,群体结构的存在会通过影响等位变异位点而影响到关联分析的准确性,易造成伪关联[65]。因此,在满足强连锁不平衡条件下构建单倍型并进行群体结构分析,获取矫正系数Q值,有利于提高关联分析的可靠性。但仅通过Q值作为矫正条件,并不能完全避免出现假阳性关联。有研究表明,以群体结构Q值和亲缘系数值共同作为协变量的混合线性模型,即利用MLM_Q+亲缘系数值模型进行关联分析,更有利于提高关联分析的准确性[8]。据此,本研究通过这种模型对两年的青枯病病情指数进行关联,在94份材料中共发现了7个单倍型(含14个位点)与青枯病抗病性相关,远多于吴超等[3]发现的6个与青枯病抗性相关的MFLP标记位点。两年间重复定到了3个单倍型与青枯病抗性相关,分别为Hap61、Hap62和Hap64,平均解释率超过10%,可为烟草青枯病抗病材料的分子标记辅助筛选提供参考。与前人[2,4-6]利用连锁分析发现烟草青枯病抗性相关的QTL位点相比,本研究所得的结果主要有以下特点。首先,本研究利用的供试材料丰富,克服了连锁分析仅利用两个亲本材料进行研究的局限性,所以本研究发现的与青枯病抗病性相关的单倍型,其适用的材料较广泛;其次,本研究作图精度较高,可以实现标记的精确定位,而连锁分析仅是将QTL定位在临近标记之间的区域;再者,本研究通过构建单倍型进行关联分析,获得的单倍型包含较丰富的信息。然而,连锁分析能检测QTL的加性效应,且能克服关联分析对稀有等位基因检测效能低的缺点[57];所以在今后的研究中,如果采用连锁分析结合关联分析的策略,扬长避短,将可提高青枯病抗性QTL分析的效率和准确性。
猜你喜欢
今日农业(2022年13期)2022-09-15 01:18:00
空间科学学报(2021年1期)2021-05-22 01:36:34
中国麻业科学(2018年6期)2018-04-09 11:22:12
环境保护与循环经济(2017年5期)2018-01-22 02:56:44
西南农业学报(2016年5期)2016-05-17 05:42:21
广西林业科学(2016年3期)2016-03-16 05:43:21
中国果菜(2016年9期)2016-03-01 01:28:44
中国蔬菜(2015年9期)2015-12-21 13:04:40
现代农业(2015年1期)2015-02-28 18:39:51
植物营养与肥料学报(2012年5期)2012-10-26 03:28:36
