
NLRs炎症体在肺感染性疾病中的作用机制研究※
2018-03-15 00:53夏金婵张小莉
中国中医药现代远程教育 2018年5期
夏金婵 张小莉
肺感染性疾病是临床上常见的疾病,包括终末气道、肺泡腔及肺间质等在内的肺实质炎症。世界卫生组织(WHO)资料显示2004年的发病率为4.29亿,成为全球第三大致死疾病。慢性阻塞性肺炎(COPD)主要是由于烟草烟雾与细菌感染引起,在许多农业国家是第四大致死原因。急性肺损伤(ALI)或急性呼吸窘迫综合征(ARDS)是有多种非心源性治病因素,例如,感染导致的毛细血管通透性增高的急性进行性低氧性呼吸功能不全或呼吸衰竭[1]。过敏性哮喘是由于过敏引起的气道炎症,导致慢性周期性的气道阻塞。长期暴露在二氧化硅、石棉或煤颗粒可导致慢性职业病尘肺。固有免疫又称非特异性免疫,是机体防御病原菌入侵的第一道防线,在肺感染性疾病中起重要的作用[2]。模式识别受体 (patter-recognition receptors,PRRs)是固有免疫受体的代表,包含不同的蛋白家族,例如,Toll样受体 (Toll-like receptors,TLRs) 定位在膜上和NOD样受体(nucleotide-binding oligomerization domainlike receptors,NLRs) 定位在胞质[3]。PRRs与病原微生物表面的病原体相关分子模式(pathogen-associated molecular patterns,PAMPs)识别和相互作用后活化下游信号通路,引起机体的炎症反应与免疫应答。最近的研究证明破坏宿主细胞的完整性与危险信号在免疫系统阻止病原菌的入侵方面也具有重要的意义。PRRs还可以与损伤相关分子模式(Damage-associated molecular patterns,DAMPs) 识别和相互作用参与无菌组织的损伤过程[4]。而且,一些PRRs还能对大颗粒物质产生反应,在尘肺病中是一个关键的炎症因子[5]。细胞因子、炎症趋化因子和黏附分子分泌失调导致的炎症反应促进了肺感染性疾病的发展。NLRs家族在人类中包含22个成员,在小鼠中更多,其结构的共同特点为:C末端为亮氨酸重复结构域(leucine-rich repeat,LRR),具有识别配体与自我调节功能;中间为NOD,可以发生自身的寡聚;N末端为可变的效应结构域,可为胱冬蛋白的募集域 (caspaserecruitment domain,CARD)、pyrin的效应结构域 (pyrin effector domain,PYD) 及细胞凋亡杆状病毒抑制蛋白重复结构域(baculoviral inhibitor of apoptosis protein repeat domain,BIR)。具有调节同型蛋白相互作用的功能,在某种程度上影响调节因子的结合及下游信号的转导。NLRs根据N端结构域的不同可以分为四个亚家族,NLRA含有反式激活子激活域 (ADs),NLRB含有BIR结构域,NLRC含有CARD,NLRP含有PYD[6]。本文就NLRs炎性体在肺感染性疾病中作用的相关研究进展做如下综述。
1 NOD1和NOD2
NOD1和NOD2是NLRs家族第一个被研究证明参与对病原菌感知的胞内蛋白,编码NOD1和NOD2的基因分别位于人类染色体7p14和16p12,在其N末端都含有CARD结构域。NOD1在许多组织与细胞中都有表达,例如:单核细胞、巨噬细胞、上皮细胞,NOD1只在一些细胞中表达,例如:树突状细胞、肺上皮细胞。NOD1和NOD2在结构上的不同主要体现在前者只有一个CARD结构域,后者有两个CARD结构域,都参与结合下游效应分子,活化核因子κβ(nuclear factor-κB,NF-κB),任何一个缺失都会影响NOD2蛋白的功能。NF-κB是天然免疫系统中重要的转录因子之一。能够调节许多炎症因子的表达,在机体抗感染过程中发挥着重要的作用。NOD受体的配体是细菌的肽聚糖,主要是N-乙酰葡糖胺 (GlcNAc,G) 和N-乙酰胞壁酸(MurNAc,M)通过短肽相互连接交替形成,在大多数革兰氏阴性与革兰氏阳性菌中NOD1识别m-DAP(LAla-g-D-Glum-diaminopimelic acid),而NOD2识别MDP(muramyl dipeptide)[7]。虽然NOD1和NOD2识别的配体结构不同,但是配体的结合都能使NOD1和NOD2蛋白中部的NOD结构域自身寡聚化形成受体复合体,受体复合体通过CARD-CARD结构域相互作用招募胞质的衔接分子受体作用蛋白-2(receptor-interacting protein-2,RIP2),泛素化的RIP2激活NF-κB。IκB激酶 (IκB kinase,IKK)复合体也参与了这一过程,IKK激酶复合体是由α、β、γ三个亚基组成,前两个为催化亚基,后一个为调节亚基,RIP2与IKKγ亚基作用激活IKK复合物,调控靶基因转录活性,参与天然免疫反应[8]。另外,在细胞内NOD1还能够活化胱冬蛋白-9(caspase-9)前体,启动细胞凋亡信号[9]。
NLRs在识别病原菌诱导细胞因子和趋化因子在病菌感染过程中起着重要的作用(图1)。研究表明敲除NOD1、NOD2或RIP2基因的小鼠感染衣原体(C.pneumoniae)、金黄色葡萄球菌(S.aureus) 或嗜肺军团菌(L.pneumophila)时肺部细胞因子和趋化因子的表达量减少,并且影响嗜中性粒细胞的向肺部的招募过程[10]。感染敲除NOD1、NOD2或RIP2基因的小鼠清除衣原体感染的能力降低,金黄色葡萄球菌感染野生型与敲除NOD2基因的小鼠时肺部的菌落数(pulmonary CFUs)并没有明显的区别,但是嗜肺军团菌感染敲除NOD1、NOD2或RIP2基因的小鼠时肺细菌负荷(pulmonary bacterial burden)提高[11]。在人的胚肾细胞HEK293中研究发现肺炎链球菌(S.pneumoniae) 诱导NF-κB活性的提高是依赖于NOD2的,利用HEK293和人的呼吸系统上皮细胞系A549研究发现在能够分泌成孔毒素溶血素的肺炎链球菌感染的情况下来自革兰氏阴性病原体流感嗜血杆菌(H.influenzae) 的肽聚糖能够激活NF-κB诱导1L-8的分泌,该过程表现出依赖NOD1的特征,流感嗜血杆菌与肺炎链球菌的成孔毒素溶血素在激活NOD1的相关性也反映出呼吸道多种微生物共存的特点[12]。分支杆菌感染敲除NOD2基因小鼠的巨噬细胞时肿瘤坏死因子α的分泌量明显减少,在人类的一个NOD2基因移码突变的纯合子个体的单核细胞中研究发现肺结核菌(M.tuberculosis)诱导的肿瘤坏死因子α的分泌量也明显减少[13]。
2 NLRC4和NAIPs
在配体的诱导下,一些NLRs蛋白能够形成异源聚合物结构成为炎性体(inflammasomes),作为平台通过CARD结构域招募胞质中的前体胱冬蛋白-1(Procaspase-1),通过溶蛋白性裂解激活caspase-1,caspase-1进而把前体1L-1β和前体1L-18激活为1L-1β和1L-18。在特定情况下caspase-1还能介导细胞凋亡。炎性体中的CARD结构域可能来自于NLRs蛋白或衔接蛋白,例如:细胞凋亡相关颗粒蛋白(apoptosis-associated speck-like protein containing a C-terminal CARD,ASC)。NLRC4和NAIP (NLR family apoptosis inducing protein)都属于NLRs蛋白家族,与其他NLRs家族的成员类似NAIPs的蛋白结构C末端为LRR,中部为NOD,N末端为BIR。配体激活后NLRC4的NOD区域与NAIP聚合形成共同形成NLRC4炎性体,激活前体caspase-1,BIR结构域参与PAMPs诱导的NLRC4寡聚化,由于NLRC4含有CARD结构域ASC衔接蛋白是否参与NLRC4炎性体的形成还有待进一步证明[14]。体外实验主要集中在对巨噬细胞中NLRC4炎性体介导的细胞因子分泌情况的分析,尽管NLRC4在上皮细胞中也有表达,但其作用机制还需要进一步研究证明[15]。在小鼠中NAIPs的同源基因有四个NAIP1、NAIP2、NAIP5与NAIP6,在人类中只研究发现一个hNAIP蛋白[16]。小鼠中NAIPs的同源基因识别不同的配体,NAIP5和NAIP6的LRR域识别鞭毛蛋白,NAIP2的LRR域识别PrgJ(typeⅢ secretion system[T3SS]needle protein),NAIP1的LRR域也识别T3SS针蛋白[17-19]。hNAIP与NAIP5识别嗜肺军团菌鞭毛蛋白,在巨噬细胞中通过NLRC4炎性体抑制细菌的生长,在人类缺失NLRC4的表皮细胞系中hNAIP能够抑制嗜肺军团菌的复制说明除了NAIP/NLRC4复合体NAIPs还有其他作用途径[20]。铜绿假单胞菌(P.aeruginosa) 通过T3SS激活NLRP4/IPAF把毒力因子分泌到宿主的胞质中,铜绿假单胞菌能够表达胞外酶U(ExoU)磷酸酶,能够抑制NLRP4/caapase-1介导的细胞因子产生,尽管临床上少于1/3的铜绿假单胞菌表达ExoU,但仍能导致严重的疾病可能与ExoU毒力因子的作用机制是不依赖于NLRP4有关[21]。肺炎克雷伯菌(K.pneumoniae)能不经过鞭毛蛋白或T3SS激活NLRP4,其激活途径还不清楚[22]。另外,在人类的巨噬细胞中堪萨斯分枝杆菌(M.kansasii)能够通过NLRP3诱导1L-1β,抑制堪萨斯分枝杆菌的生长[23]。细胞凋亡是依赖caspase-1的程序性细胞死亡过程,位于炎性体的下游,激活的caspase-1在细胞膜上形成气孔,允许阳离子(Ca2+)穿过细胞膜进入胞质,经过一系列细胞生理过程最终导致细胞死亡[24]。炎性体也不是总导致细胞凋亡,肺炎克雷伯菌在小鼠的巨噬细胞与中性粒细胞中经NLRC4激活1L-1β,但并没有观察到细胞凋亡现象[25]。相反,嗜肺军团菌与铜绿假单胞菌可经过NLRC4诱导细胞凋亡[26]。细胞的自噬作用或许参与了细胞凋亡过程,当嗜肺军团菌感染小鼠的巨噬细胞时NLRC4与NAIP5参与自噬体的形成过程,细胞的自噬作用抑制细胞凋亡过程[27]。
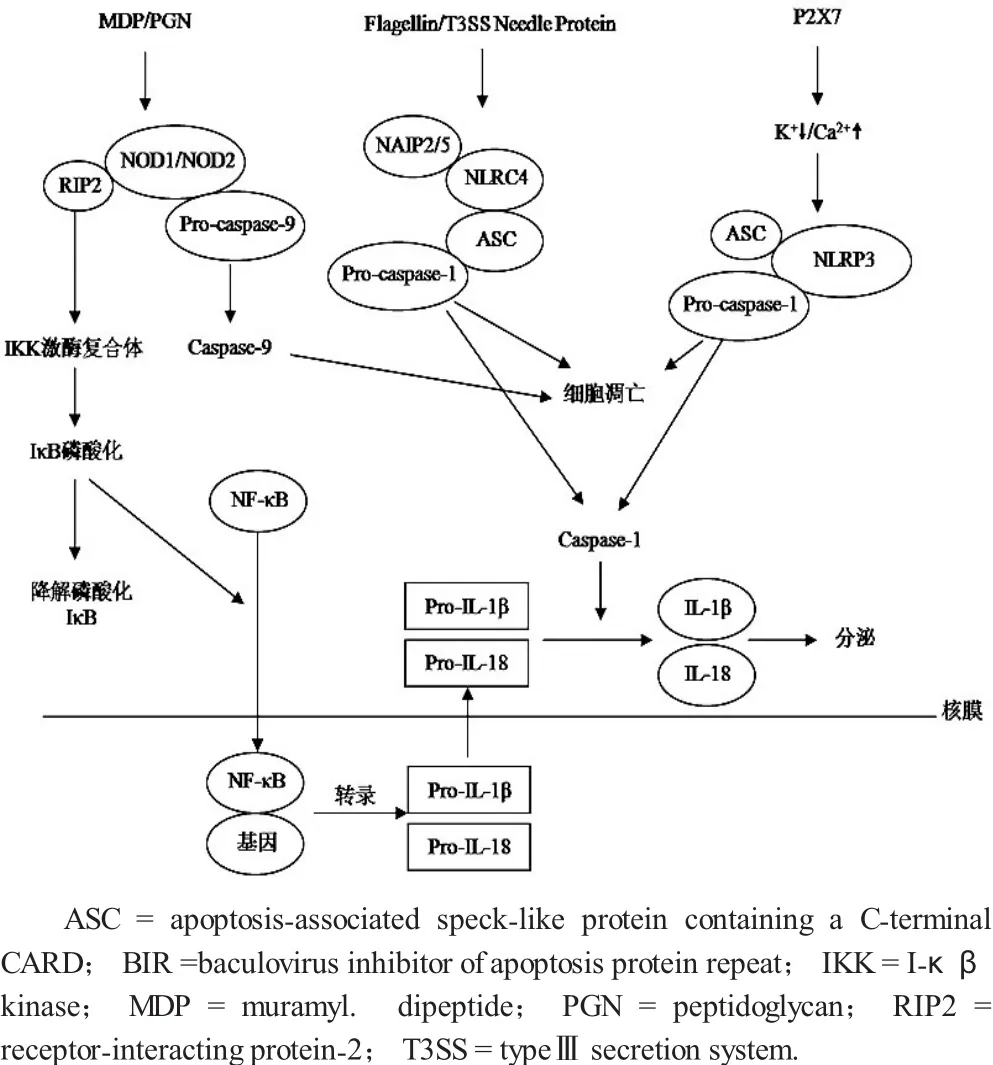
图1 细胞中NLRs信号转导途径
胞质中的NLRs与病原微生物表面的病原体相关分子模式 (pathogen-associated molecular patterns,PAMPs)识别和相互作用后激活下游促炎症信号级联通路,引起机体的防御或炎症反应。
3 NLRP3
NLRP3不仅在人和小鼠的巨噬细胞中表达,而且在人和小鼠呼吸道上皮细胞中NLRP3炎性体参与细菌引起的炎症反应过程[28-31]。NLRP3是NLRs家族中NLRP亚家族的成员,可被PAMPs和DAMPs激活,前者包括病毒与细菌(如流感病毒、金黄色葡萄球菌、结合分枝杆菌、衣原体)等,后者包括组织损伤、应激、感染等情况下宿主细胞释放的活性氧、尿酸盐结晶、ATP以及外源性物质二氧化硅、石棉等,然后在相关蛋白与酶的作用下形成NLRP3炎性体。NLRP3炎性体是由NLRP3蛋白、ASC、caspase-1组成。NLRP3炎症复合体可以活化Pro-caspase-1,激活的caspase-1可以通过剪切的方式活化炎症因子Pro-1L-1β及Pro-1L-18,从而使大量成熟的1L-1β及1L-18得以释放,参与机体的固有免疫反应,NLRP3通过N末端的PYD结构域和ASC的PYD结构域相互作用,招募激活前体胱冬蛋白-1(Procaspase-1),ASC是参与凋亡的含有195个氨基酸残基的接头蛋白,N末端是PYD结构域,可与NLRP3的N末端PYD结构域作用调节CARD结构域的寡聚状态,ASC的C末端为CARD结构域受上游信号分子调节寡聚化[32-34]。NLRP3炎性体的激活需要两种信号分子,首先通过TLR激活NF-κB诱导NLRP3的表达,当胞内NLRP3的表达量达到一定阈值后在NLRP3配体的作用下形成炎性体,尽管其他的炎性体可能不受TLR的影响,但是TLR信号确实能够增加胞内Pro-1L-1β的表达量及成熟的1L-1β的释放[35]。通常情况下NLRP3炎性体在识别病原体或机体自身危险信号前处于自身抑制状态,对其激活的机制尚未完全清楚,目前发现NLRP3的激活方式有三种:(1)细胞外的三磷酸腺苷激活细胞膜受体P2X7,激活的P2X7介导K+的外流与Ca2+内流,同时Pannexin-1在细胞膜上形成小孔,配体内流激活NLRP3,细胞在许多情况下都能表现出K+的外流,例如:膜通透性的改变、膜上小孔的形成,溶酶体与线粒体的损伤等;(2) 活性氧(ROS) 参与NLRP3炎性体的激活,研究发现NLRP3炎性体的激活剂可以诱导ROS的产生,ROS的抑制剂可以抑制炎性体的激活[36];(3)当硅、石英、胆固醇结晶等晶体类物质被细胞吞噬后形成内吞泡,与溶酶体结合后破坏溶酶体,从溶酶体中释放一些酶类物质如组织蛋白酶B,可以直接或间接激活NLRP3炎性体[37]。
NLRP3炎性体能够识别危险信号,释放炎症因子,引起炎症反应,是目前研究相对较多的一个炎性体,参与多种肺部疾病的发生。金黄色葡萄球菌能够引起炎症反应,导致肺组织的坏死,甲氧西林金黄色葡萄球菌的毒力因子α溶血素通过活化NLRP3炎性体产生大量细胞因子1L-1β与1L-18,导致细胞程序化死亡,NLRP3缺失的小鼠感染金黄色葡萄球菌导致的肺炎程度较轻。肺炎链球菌溶血素与其他细菌的成孔毒素,例如:链球菌溶血素 O(Streptococcus pyogenes)、α溶血素(S.aureus),也能诱导NLRP3炎性体的形成。在人类和小鼠的巨噬细胞中能够分泌成孔毒素溶血素的肺炎链球菌 (S.pneumoniae) 能够经过NLRP3诱导1L-1β,而且激活NLRP3可对抗对肺炎链球菌感染小鼠[38]。研究还发现NLRP3炎性体不仅在防御早期的肺炎链球菌肺炎免疫反应中发挥着重要作用,还参与A型流感病毒性肺炎、结核分枝杆菌的炎症反应[39-41]。临床上由类鼻疽伯克菌引起的类鼻疽病导致的肺部感染很常见,在类鼻疽病小鼠模型中巨噬细胞与树突状细胞中1L-1β与1L-18的表达明显增高,NLRP3缺失的小鼠对类鼻疽伯克菌的敏感性增强[42]。在衣原体肺炎小鼠模型中肺部通过NLRP3炎性体产生大量1L-1β,caspase-1缺失的小鼠1L-1β的产生延迟,肺部清除细菌的能力降低,死亡率增高[43]。慢性阻塞性肺炎主要是由于烟草烟雾与细菌感染引起的多因子疾病,长期慢性刺激导致气道重塑、黏液纤毛清除能力降低或肺实质受损。CS诱导尿酸与焦磷酸钙的形成,通过NLRP3炎性体激活caspase-1[44]。目前越来越多的研究表明NLRP3炎性体在COPD与哮喘的慢性炎症中发挥作用。研究表明caspase-1或NLRP3缺失的COPD小鼠肺组织匀浆、BALF和诱导痰中1L-1β大量减少,1L-1β是1L-1受体的重要配体,敲除1L-1受体小鼠肺组织中巨噬细胞、中性粒细胞、树突状细胞及活化的CD4+和CD8+T细胞等炎症细胞明显减少[45]。在小鼠哮喘模型的巨噬细胞和树突状细胞中血清淀粉样蛋白A通过激活NLRP3炎性体分泌1L-1β[46]。
细菌性肺疾病发病率逐年提高,肺部对细菌的固有免疫反应是一把双刃剑,受损的反应可能会导致危及生命的感染,而一个不受控制的反应可能会导致危及生命的炎症性疾病。尽管NLRs在该过程起到重要的作用,但还有许多未知问题。NLRs家族的新成员、配体及下游的调控因子还在不断被发现,TLRs与NLRs受体,以及与细胞的自我吞噬之间相互联系的分子调控的机制也是一个新的研究方向。针对NLRs炎症复合体的靶向治疗可能成为肺感染性疾病防治的方法之一,在动物模型中利用caspase-1、1L-1β和1L-1受体的拮抗剂、尿酸抑制剂、尿酸酶、别嘌呤醇治疗TLRs介导的炎症已取得了很大的成功[47]。尽管已经证明NLRs参与了肺感染性疾病及慢性炎症的发生过程,然而,其参与的分子机制尚未完全明白,进一步研究其激活或调控机制为肺感染性疾病的治疗提供新的思路与手段。
[1]MatthayMA,Zemans RL.The Acute Respiratory Distress Syndrome:Pathogen-Esisand Treatment[J].Annu Rev Pathol,2011,6:147-163.
[2]ParkerD,PrinceA.InnateImmunityintheRespiratoryEpithelium[J].AmJRespir Cell Mol Biol,2011,45:189-201.
[3]Lamkanfi M,DixitVM.Inflammasomes and Thei Rroles in Health and Disease[J].Annu Rev Cell Dev Biol,2012,28:137-161.
[4]RockKL,LatzE,OntiverosF,KonoH.The Sterile Inflammatory Response[J].Annu Rev Immunol,2010,28:321-342.
[5]CasselSL,EisenbarthSC,IyerSS,et al.The Nalp3 Inflammasome is Essential for the Development of Silicosis[J].Proc Natl Acad Sci USA 2008,105:9035-9040.
[6]Franchi L,Warner N,Viani K,et al.Function of Nod-like receptors in microbial recognition and host defense[J].Immunol Rev 2009,227:106-128.
[7]Correa RG,Milutinovic S,Reed JC.Rolesof NOD1(NLRC1)and NOD2(NLRC2)in innate immunity and inflammatory diseases[J].Biosci Rep 2012,32:597-608.
[8]Inohara N,Koseki T,Lin J,et al.An induced proximity model for Nfkappa B activation in the Nod1 RICK and RIPsignaling pathways[J].JBiol Chem,2000,275:27823-27831.
[9]Yoo NJ,Park WS,Kim SY,et al.Nod 1,a CARD protein,enhances prointerleukin-1beta processing through the interaction with pro-caspase-1[J].Biochem Biophys Res Commun,2002,299:652-658.
[10]Berrington WR,Iyer R,Wells RD,etal.NOD1 and NOD2 regulation of pulmonaryinnateimmunitytoLegionella pneumophila[J].Eur JImmunol 2010;40:3519-3527.
[11]Shimada K,Chen S,Dempsey PW,et al.The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniaelunginfection[J].PLoSPathog2009,5:e1000379.
[12]Ratner AJ,Aguilar JL,Shchepetov M,et al.Nod1mediates cytoplasmic sensing of combinations of extracellular bacteria[J].Cell Microbiol 2007,9:1343-1351.
[13]Ferwerda G,Girardin SE,Kullberg BJ,et al.NOD2 and Toll-like receptors arenonredundant recognition systemsof Mycobacteriumtuberculosis[J].PLoS Pathog2005,1:279-285.
[14]Kofoed EM,Vance RE.NAIPs:building an innate immune barrier against bacterial pathogens.NAIPs function as sensors that initiate innate immunity by detectionof bacterial proteinsin thehostcell cytosol[J].Bioessays2012,34:589-598.
[15]Hu B,Elinav E,Huber S,et al.Inflammation-induced tumorigenesis in the colonisregulatedbycaspase-1and NLRC4[J].Proc Natl Acad SciUSA 2010,107:21635-21640.
[16]Endrizzi MG,Hadinoto V,Growney JD,et al.Genomic sequence analysis of the mouse Naip gene array[J].Genome Res2000,10:1095-1102.
[17]Kofoed EM,Vance RE.Innate immune recognition of bacterial ligands by NAIPsdeterminesinflammasome specificity[J].Nature2011,477:592-595.
[18]Zhao Y,Yang J,Shi J,et al.The NLRC4 inflammasomereceptorsfor bacte rial flagellin and type IIIsecretion apparatus[J].Nature 2011,477:596-600.
[19]Rayamajhi M,Zak DE,Chavarria-Smith J,etal.Cuttingedge:mouse NAIP1 detects the type IIIsecretion system needle protein[J].JImmunol 2013,191:3986-3989.
[20]Vinzing M,Eitel J,Lippmann J,et al.NAIP and Ipaf control Legionella pneumophila replication in human cells[J].JImmunol 2008,180:6808-6815.
[21]Patankar YR,Lovewell RR,Poynter ME,et al.Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa[J].Infect Immun 2013,81:2043-2052.
[22]Patankar YR,Lovewell RR,Poynter ME,et al.Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa[J].Infect Immun 2013;81:2043-2052.
[23]Chen CC,Tsai SH,Lu CC,et al.Activation of an NLRP3 inflammasome restricts Mycobacteriumkansasiiinfection[J].PLoSOne2012,7:e36292.
[24]LaRock CN,Cookson BT.Burning down the house:cellular actions during pyroptosis[J].PLoSPathog2013,9:e1003793.
[25]Cai S,Batra S,Wakamatsu N,et al.NLRC4 inflammasome-mediated production of IL-1b modulates mucosal immunity in the lung against gramnegativebacterial infection[J].JImmunol 2012,188:5623-5635.
[26]Miao EA,Leaf IA,Treuting PM,et al.Caspase-1 induced pyroptosis is an innate immune effector mechanism against intracellular bacteria[J].Nat Immunol 2010,11:1136-1142.
[27]Amer AO,Swanson MS.Autophagy is an immediate macrophage response to Legionellapneumophila[J].Cell Microbiol 2005,7:765-778.
[28]Rotta detto Loria J,Rohmann K,Droemann D,et al.Nontypeable Haemophilus Influenzae Infection Upregulatesthe NLRP3 inflammasome and leadstocaspase-1 dependent secretion of interleukin-1b—apossiblepathway of exacerbationsin COPD[J].PLoSOne2013,8:e66818.
[29]Jiang L,Fei D,Gong R,et al.CORM-2 inhibits TXNIP/NLRP3 inflammasomepathway in LPS-induced acutelunginjury[J].Inflamm Res.2016 Jul 13.[Epub ahead of print]
[30]Yin N,Peng Z,Li B,et al.Isoflurane attenuates lipopolysaccharide-induced acute lung injury by inhibiting ROS-mediated NLRP3 inflammasome activation[J].Am JTransl Res.2016 May 15;8(5):2033-2046.
[31]Wang Y,Kong H,Zeng X,et al.Activation of NLRP3 inflammasome enhances theproliferation and migrationof A549lungcancer cells[J].Oncol Rep.2016 Apr,35(4):2053-2064.
[32]Pétrilli V,Dostert C,Muruve DA,et al.Theinflammasome:adanger sensing complex triggering innate immunity[J].Curr Opin Immunol,2007 ,19 :615-622.
[33]Luna-Gomes T,Santana PT,Coutinho-Silva R.Silica-induced inflammasome activation in macrophages:role of ATPand P2X7 receptor[J].Immunobi ology.2015 Sep,220(9):1101-1106.
[34]Hosseinian N,Cho Y1,Lockey RF,Kolliputi N.The role of the NLRP3 inflammaso me in pulmonary diseases[J].Ther Adv Respir Dis.2015 Aug,9(4):188-97.
[35]Jin C,Flavell RA.Molecular mechanismof NLRP3inflammasomeactivation[J].JClin Immunol 2010,30:628-631.
[36]Munoz-Planillo R,Kuffa P,Martinez-Colon G,et al.K1 efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter[J].Immunity 2013;38:1142-1153.
[37]HalleA,HornungV,Petzold GC,etal.TheNALP3inflammasomeisinvolved in the innateimmuneresponsetoamyloid-beta[J].Nat Immunol,2008,9:857-865.
[38]Witzenrath M,Pache F,Lorenz D,et al.The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia[J].JImmunol 2011,187:434-440.
[39]Craven RR,Gao X,Allen IC,et al.Staphylococcus aureus alphahemolysin activates the NLRP3inflammasome in human and mouse monocytic cells[J].PLoSOne,2009,4:e7446.
[40]Thomas PG ,Dash P ,Aldridge JR Jr,et al.The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1[J].Immunity,2009,30:566-575.
[41]Dorhoi A,Nouailles G,Jrg S,et al.Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis[J].Eur JImmunol,2012,42:374-384.
[42]Ceballos-Olvera I,Sahoo M,Miller MA,et al.Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1βisdeleterious[J].PLoSPathog,2011,7:e1002452.
[43]Shimada K,Crother TR,Karlin J,et al.Caspase-1 dependent IL-1β secretion is critical for host defense in a mouse model of Chlamydia pneumoniae lunginfection[J].PLoSOne,2011,6:e21477.
[44]Pauwels NS,Bracke KR,Dupont LL,et al.Role of IL-1a and the Nlrp3/caspase-1/IL-1b axisin cigarette smoke-induced pulmonary inflammation and COPD[J].Eur Respir J2011,38:1019-1028.
[45]Pauwels NS,Bracke KR,Dupont LL,et al.Role of IL 1α and the Nlrp3/caspase 1/IL 1βaxis in cigarette smokeinduced pulmonary inflammation and COPD[J].Eur Respir J,2011,38:1019-1028.
[46]Ather JL,Ckless K,Martin R,et al.Serum amyloid A activates the NLRP3 inflammasome and promotes Th1 7 allergic asthma in mice[J].JImmunol,2011,187 :64-73.
[47]Kuipers MT,AslamiH,Janczy JR,etal.Ventilatorinduced lunginjuryismediated bytheNLRP3inflam masome[J].Anesthesiology2012,116:1104-1115.
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
昆明医科大学学报(2022年2期)2022-03-29
中老年保健(2021年3期)2021-12-03
中华养生保健(2020年9期)2021-01-18
实用肿瘤学杂志(2020年4期)2020-12-08
世界农药(2019年2期)2019-07-13
中华老年多器官疾病杂志(2016年9期)2016-04-28
医学研究杂志(2015年2期)2015-06-10
中国老年学杂志(2015年9期)2015-01-31
中国中医药现代远程教育(2014年15期)2014-03-01
