
Gilbert综合征2例报道
2018-03-13 06:44张传朓张继平
实用肝脏病杂志 2018年1期
张传朓,张继平
Gilbert综合征是一种遗传性非结合性高胆红素升高性疾病,是临床上最为常见的一种先天性黄疸。近年,从组织病理学和基因水平诊断该病增多。现就近期我院经肝组织病理学和基因检测确诊的2例患者的临床资料进行分析,并复习了有关文献,现报道如下。
1 病例摘要
例1女,36岁。因“反复眼黄、尿黄、皮肤黄36年”于2016年6月15日入院。缘于出生后即被发现眼黄、皮肤黄,尿黄,饮食等日常生活如常,无皮肤瘙痒。生长发育正常,尿黄因过度劳累或熬夜后加重,经休息后缓解。多次在多家医院就诊,检查提示总胆红素(TBIL)波动于120~150μmol/L,谷丙转氨酶(ALT)正常,未明确诊断,常规治疗无好转。10年前,妊娠时血清TBIL升高至250 μmol/L,分娩后恢复至以前水平。患者亲属中无类似症状者。体检:巩膜、皮肤中度黄染,未见肝掌、蜘蛛痣,腹平坦、肝脾未触及,肝区无叩痛。化验:TBIL 153.9μmol/L,IBIL67.0 μmol/L,血清 HBsAg、HBV DNA、抗 -HCV、HCV RNA、抗 -HIV、抗 HAV-IgM、抗HEV-IgM、抗CMV-IgM、抗EBV-IgM、常见自身抗体均阴性。骨髓穿刺检查、血常规、尿常规、血清甲胎蛋白、凝血功能和其他生化指标均无明显异常。上腹部CT检查无明显异常。肝穿刺活组织检查标本送广州金域医学检验中心病理科检查,发现肝小叶结构存在,部分汇管区轻微扩大,未见炎性细胞浸润。区域性毛细胆管轻度扩张,淤胆性色素颗粒有围绕毛细胆管现象(图1~3),将外周血送往北京宏微特斯生物科技有限公司行基因检测,发现患者尿苷葡萄糖醛酸转移酶基因突变点位于启动子上游PBREM-3263(-3279)以编码区外显子EX0N1上的GGAAGA Gly71Arg(表1),最终确诊为Gilbert综合征。

图1 肝组织病理学表现 光镜下,肝小叶结构存在,部分汇管区轻微扩大,未见炎性细胞浸润(HE,200×)

图2 肝组织病理学表现 光镜下,肝细胞轻微水肿,部分略呈毛玻璃样,可见糖原核肝细胞,中央静脉周围肝细胞内少量淤胆性色素颗粒(HE,200×)

图3 肝组织超微结构改变 电镜下,肝细胞肿胀,部分滑面内质网轻度扩张。部分肝细胞内可见少量淤胆性色素颗粒,区域性毛细胆管轻度扩张,淤胆性色素颗粒有围绕毛细胆管现象。肝窦内皮细胞肿胀,窦周偶见星状细胞。肝细胞间及Disse间隙未见束状胶原纤维沉积

表1 基因检测图谱
例2男,30岁。因“眼黄、尿黄、皮肤黄7年”于2016年5月30日就诊我院。缘于2009年春无明显诱因下出现眼黄、尿黄、皮肤黄,血清TBIL最高时达75.0μmol/L,尿黄可因过度劳累、饮酒或熬夜后加深,经休息后减轻。就诊我院时 TBIL 64.3 μmol/L,IBIL 45.2 μmol/L,MRCP、腹部彩超提示:脾肿大(厚约62 mm),其他发病情况类似例1。患者为独子,父母及其他亲属无类似情况。肝穿刺组织病理学检查发现肝小叶结构存在,未见炎性反应,中央静脉周围肝细胞内少量淤胆性色素颗粒,普鲁士蓝染色可见Kupffer细胞内少许铁颗粒,肝细胞内可见多少不一的淤胆性色素颗粒,区域性毛细胆管扩张(图4~7)。基因检测发现患者基因突变点位于启动子上游PBREM-3263(-3279,表2)。最终确诊为Gilbert综合征。

图4 肝组织病理学表现 光镜下,肝小叶结构存在,汇管区未见扩大,未见炎性反应(HE,200)
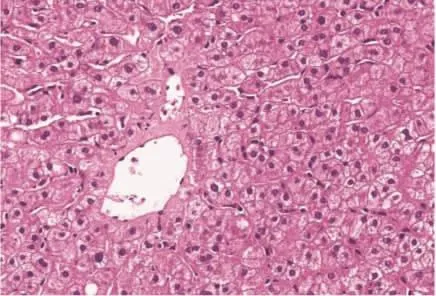
图5 肝组织病理学表现 肝细胞损伤不明显,中央静脉周围肝细胞内少量淤胆性色素颗粒(HE,200×)

图6 肝组织病理学表现 可见肝细胞内少量淤胆性色素颗粒,Kupffer细胞内少许铁颗粒(普鲁士蓝染色,200×)

图7 肝组织超微结构改变 肝细胞肿胀,部分肝细胞间隙稍增宽,少数肝细胞间可见微绒毛结构,内质网明显扩张,个别肝细胞内偶见髓鞘样小体,线粒体肿胀、增大,增大的线粒体内可见结晶体。肝细胞内可见多少不一的淤胆性色素颗粒,区域性毛细胆管扩张。肝窦内皮细胞肿胀,偶见星状细胞。区域性Disse间隙有少量束状胶原纤维沉积

表2 基因检测图谱
2 讨论
Gilbert综合征于1901年由法国学者Gilbert和Lereboulle首次报道,以慢性、间歇性、轻度高非结合型胆红素升高为特征,无明显溶血依据和其他肝脏疾病表现。本病在西方国家很常见,据称发病率达2%~19%[1-3]。我国尚缺乏大数据统计资料,但从临床实践看,本病在我国并不少见。本病多于15~30岁发病,也有自小或成年期发病,男性多见,男女比例为4:1[1-3],多有家族史。本病起病隐匿,多无明显症状,常因眼黄或体检意外发现胆红素升高,少数患者可能有轻度乏力、纳差或肝区不适,其程度与黄疸深度无关,可能为焦虑等精神因素所致。剧烈运动、劳累、饥饿、创伤、感染,或者妊娠可能为诱因导致黄疸加重,除黄疸外,无其他阳性体征。
Gilbert综合征肝组织病变轻微,肝小叶结构基本保存,肝板排列整齐。汇管区及小叶内少见炎症细胞浸润,汇管区少见扩大及纤维化,中央静脉周围肝细胞内可见较细的棕褐色色素颗粒沉积。电镜下,可见肝细胞肿胀,内质网明显扩张,线粒体肿胀、增大。肝细胞内可见多少不一的淤胆性色素颗粒,区域性毛细胆管扩张,淤胆性颗粒多分布在毛细胆管周围。肝窦内皮细胞肿胀,偶见星状细胞。区域性Disse间隙有少量束状胶原纤维沉积。本文2例患者肝组织病理学表现符合以上特征。
Gilbert综合征是一个遗传性疾病,遗传方式为常染色体隐性遗传和常染色体显性遗传(不完全外显)。本病的发病原因是位于染色体2q37位点的胆红素尿苷葡萄糖醛酸转移酶基因(UTG1A1基因)发生缺陷,导致B-UGT表达下降。随着分子生物学技术的发展,对Gilbert综合征人群UGT1酶活性低下遗传背景已有较多的了解,Sampietro et al[4]研究显示UTG1酶基因在启动子区的TATA-BOX发生变异。Clarke[5]进一步研究表明是UGT1外显子上游TATA启动子区一个TA碱基对的插入。Jansen P[6]经过对UGT1启动子区基因测序,发现正常人群启动子区基因为纯合子,含6/6TA重复,Gilbert综合征突变为7/7TA重复的杂合子。Debinski H[7]采用免疫组化检测技术在蛋白水平显示肝小叶内UGT1酶染色强度比正常对照明显降低。通过遗传筛查寻找出致病基因位点,针对临床不明原因的黄疸患者进行基因筛查,可确诊本病,同时可与其他黄疸性疾病进行鉴别,对该病的预后判断有重要意义。
Gilbert综合征应与以下常见病鉴别。(1)Dubin-Johnson综合征:除黄疸外,大多数伴有腹痛、乏力、恶心、呕吐等症状,肝肿大、肝区压痛。口服胆囊造影常不显影或很淡。肝组织病理学表现可见小叶结构正常,唯一的特征为肝细胞内有很多大小不等的棕色素颗粒,以小叶区最为显著,此色素被认为是脂褐素和黑色素,系肾上腺素代谢物的多聚体。该病也是一种遗传性疾病,染色体10q23-q24上MRP2基因有多种变异;(2)慢性胆汁淤积型药物性肝损伤:有药物接触史,黄疸呈持续性,病理学上可见胆管损伤,伴或不伴胆管硬化或缺失,汇管区周围胆盐沉积,铜沉积,汇管区纤维化[8];(3)原发性胆汁性肝硬化:多见于中老年女性,常伴疲乏和皮肤瘙痒,病理学表现为进行性、非化脓性、破坏性肝内小胆管炎,可发展至肝硬化,血清抗线粒体抗体(AMA)阳性,尤其是AMA-M2亚型阳性对本病诊断具有很高的敏感性和特异性[9];(4)原发性硬化性胆管炎:多见于中年男性,部分合并溃疡性结肠炎,胆管造影典型表现为胆管不规则、多发性局部狭窄和扩张,胆道明显狭窄伴正常扩张段形成串珠样改变[10]。
Gilbert综合征预后良好,不影响生活和工作,对寿命亦无影响[11],多无需治疗。本病早期诊断更有意义,可避免被误诊为肝细胞性黄疸、梗阻性黄疸或溶血性黄疸等,有些患者甚至辗转多家医院治疗,增加患者经济及心理负担[12-14]。应给患者进行健康教育,消除心理障碍,注意避免可能的诱因,加重病情。
[1]D'Apolito M,Marrone A,Servedio V,et al.Seven novel mutations of the UGT1A1 gene in patients with unconjugated hyperbilirubinemia.Haematologica,2007,92:133-134.
[2]Ostanek B,Furlan D,Mavec T,et al.UGT1A1 (TA)n promoter polymorphism—a new case of a(TA)8 allele in Caucasians.Blood Cells Mol Dis,2007,38:78-82.
[3]Teng HC,Huang MJ,Tang KS,et al.Combined UGT1A1 and UGT1A7 variant alleles are associated with increased risk of Gilbert's syndrome in Taiwanese adults.Clin Genet,2007,72:321-328.
[4]Sampietro M,Lupica L,Perrero L,et al.TATA-box promoter mutant in the promoter of UDP-glucuronosyltransferase gene in Italian patients with Gilbert's syndrome.Ital J Gastroenterol,1998,30:194-198.
[5]Clarke DJ,Moghrabi N,Monaghan G,et al.Genetic defects of the UDP-glucuronosyltransferase-1 (UGT1) gene thatcause familial non-haemolytic unconjugated hyperbilirubinaemias.Clin Chim Acta,1997,299:63-74.
[6]Jansen PL1,Bosma PJ,Bakker C,et al.Persistent unconjugated hyperbilirubinemia after liver transplantation due to an abnormal bilirubin UDP-glucuronosyl-transferase gene promotor sequence in the donor.J Hepatol,1997,27:1-5.
[7]Deinski HS,Lee CS,Dhillen AP,et al.UDP-glucUronosyltransferase in Gilbert's syndrome.Pathology,1996,28:238-241.
[8]Kleiner DE.Histopathological evaluation of drug-induced liver disease.Academic Press,2013:241-261.
[9]中华医学会肝病学分会、消化病学分会、感染病学分会.胆汁淤积性肝病诊断和治疗共识(2015).实用肝脏病杂志,2016,19(6):Ⅰ-ⅩⅠ.
[10]中华医学会肝病学分会、消化病学分会、感染病学分会.原发性硬化性胆管炎诊断和治疗专家共识(2015).中华肝脏病杂志,2016,24(1):14-22.
[11]Strassburg CP. Pharmacogenetics of Gilbert's syndrome.Pharmacogenomics,2008,9(6):703-715.
[12]张玉喜,马晓瑞,马国仁.Gilbert综合征21例临床分析.宁夏医学杂志,2010,32:634-635.
[13]孙艳玲,赵景民,辛绍杰,等.几种主要的先天性胆红素代谢障碍性肝病的临床及病理研究.中华传染病信息,2008,21(5):287-290.
[14]彭向欣,王泰龄.16例Gilbert综合征患者的临床、病理特征和基因分析.中华肝脏病杂志,2008,16(5):372-374.
猜你喜欢
现代仪器与医疗(2022年3期)2022-08-12
肝博士(2022年3期)2022-06-30
昆明医科大学学报(2021年5期)2021-07-22
中国毕业后医学教育(2020年4期)2020-12-06
陶瓷学报(2019年6期)2019-10-27
真空与低温(2019年3期)2019-07-04
益寿宝典(2018年14期)2018-01-27
中国卫生标准管理(2015年18期)2016-01-20
中国卫生标准管理(2015年16期)2016-01-20
中国卫生标准管理(2015年3期)2016-01-14
