
表面接枝亲水性聚合物刷磁性微球的制备及其对蜂蜜中四环素类抗生素残留的磁分散固相萃取
2018-03-05 01:06:09周少丹唐嘉琦吕运开
色谱 2018年2期
周少丹, 唐嘉琦, 贾 博, 吕运开
(河北大学化学与环境科学学院, 河北省分析科学技术重点实验室, 河北 保定 071002)
四环素类抗生素(tetracycline antibiotics, TCs)具有广谱抗菌性,在畜禽养殖业中常被作为饲料添加剂用于预防和治疗动物疾病。近年来,畜禽养殖业中抗生素的不合理使用和滥用现象十分普遍,而可食用性动物组织中抗生素及其代谢物残留对人类健康存在潜在危害。摄入过量含抗生素的动物源食品会使人发生过敏反应,产生耐药性。因此,抗生素残留问题引发了国内外的广泛关注,美国食品药品监督管理局(FDA)[1]、欧盟[2]和中国农业部[3]均规定TCs在动物源食品中的最大残留限量(MRL)为0.10 mg/kg。
食品中四环素类抗生素残留的主要检测方法有色谱法[4-6]及色谱-质谱联用法[7,8],而样品前处理步骤是影响检测灵敏度和准确度的关键环节。目前检测TCs残留的前处理方法主要是固相萃取技术[5,6,9]。尽管固相萃取技术具有溶剂用量小、操作简便等优点,但仍存在萃取过程繁琐、耗时长等缺点[9]。磁分散固相萃取(magnetic dispersion solid phase extraction, MDSPE)技术[9-11]因其简便快速的优点得到了迅速发展,其研究核心是功能化磁性微球(MMs)的制备。在前期研究[12-15]中发现亲水性磁性微球可以使微球表面形成一个有效的亲水层,提高了水相体系的相容性和样品净化能力(主要是表面水合层抑制了生物大分子的吸附),但表面接枝层的厚度仍影响实际样品的净化效果。为了提高净化效果,本工作在磁性微球表面接枝了双功能聚合物刷,合成了亲水性聚合物刷磁性微球(HMMs),以解决亲水层厚度低的问题,实现了尺寸排阻生物大分子。
目前合成亲水性聚合物刷磁性微球常用的方法有可逆加成-断裂链转移聚合法(RAFT)[16]、原子转移自由基聚合法(ATRP)[17]和电子转移活化再生催化剂原子转移自由基聚合法(ARGET-ATRP)[18]。RAFT适用于聚合较不活泼的单体,但采用RAFT聚合时难以控制反应程度与产物的相对分子质量;ATRP适用的单体较多,可进行本体、溶液、乳液、悬浮等聚合,但存在催化剂易被氧化、用量大、聚合后难脱除等问题;ARGET-ATRP既可以控制聚合物刷的相对分子质量,又解决了ATRP需严格无氧的苛刻条件,引起了研究者的极大兴趣[18]。
本文采用ARGET-ATRP制备了内层为聚碱类聚合物、外层为亲水性聚合物刷的磁性微球,并利用磁分散固相萃取-高效液相色谱法(HPLC)测定蜂蜜中的盐酸四环素(tetracycline hydrochloride, TC)、盐酸金霉素(chlortetracycline hydrochloride, CTC)和盐酸强力霉素(doxycycline hydrochloride, DC)残留。通过优化MDSPE萃取条件(上样溶液的pH、萃取时间、淋洗液和洗脱剂),发展了一种快速分离、富集、检测蜂蜜中四环素类抗生素残留的方法。
1 实验部分
1.1 仪器与试剂
LC-20A高效液相色谱仪、TGA-50热重分析仪、UV-3600紫外可见分光光度计(日本岛津公司); JEM-100SX透射电子显微镜(日本电气股份有限公司); Nicolet iS10傅里叶变换红外光谱仪(美国赛默飞世尔科技公司)。
四水合二氯化亚铁(FeCl254H2O)、六水合三氯化铁(FeCl356H2O)、乙醇、氢氧化钠、油酸、正硅酸乙酯(TEOS)、氨水、盐酸、甲苯、二氯甲烷和三乙胺均购自天津市科密欧化学试剂有限公司;甲醇、乙酸、乙酸乙酯、草酸、乙腈、二水乙二胺四乙酸二钠(Na2EDTA52H2O)、柠檬酸、磷酸氢二钠、磷酸和硼酸均购自天津市东华试剂厂;四氢呋喃购自天津市进丰化工有限公司;考马斯亮蓝G-250购自北京欣惠泽莫科技有限公司;2-溴异丁酰溴、溶菌酶(lysozyme, LZM)和鱼精蛋白(protamine, PRM)均购自上海阿拉丁生化科技股份有限公司;氢化钙(CaH2)、溴化铜(CuBr2)、甲基丙烯酸二乙氨基乙酯(DEAEMA)、聚(乙二醇)甲基丙烯酸酯(poly(ethylene glycol) methacrylate, PEGMA,Mr=400)、异辛酸亚锡(Sn(Oct)2)均购自上海麦克林生化有限公司;五甲基二乙烯基三胺(N,N,N′,N″,N″-pentamethyl diethylene triamine, PMDETA)购自玛雅试剂;3-氨丙基三乙氧基硅烷((3-amino propyl) triethoxy silane, APTES)购自上海邦成化工有限公司;TC、CTC和DC均购自美国Fluka公司。实验所用其他试剂均为分析纯。蜂蜜购自当地超市。
1.2 溶液的配制
Mcllvaine缓冲溶液(pH 4.00±0.05):将6.45 g柠檬酸、13.80 g磷酸氢二钠、18.60 g二水乙二胺四乙酸二钠盐用水溶解并定容至500 mL。
Na2EDTA-Mcllvaine缓冲溶液(0.10 mol/L):称取60.50 g二水乙二胺四乙酸二钠,加入1 625.0 mL Mcllvaine缓冲溶液,溶解,摇匀。
伯瑞坦-罗宾森(Britton-Robinson, B-R)缓冲溶液Ⅰ(pH 6.00±0.10):在100.0 mL浓度均为0.04 mol/L的磷酸、乙酸和硼酸混合溶液中加入42.5 mL 0.20 mol/L NaOH溶液。
B-R缓冲溶液Ⅱ(pH 10.00±0.10):在100.0 mL浓度均为0.04 mol/L的磷酸、乙酸和硼酸混合溶液中加入77.5 mL 0.20 mol/L NaOH溶液。
考马斯亮蓝试剂的配制:称取10 mg考马斯亮蓝G-250溶于5 mL 95%(v/v)乙醇水溶液中,然后与10 mL 85%(v/v)磷酸水溶液混合,用蒸馏水定容至100 mL,滤纸过滤,待用。
TCs标准储备液(1.00 g/L):准确称取0.01 g TC、CTC和DC标准品,加入少量甲醇溶解,然后转移至10 mL棕色容量瓶中,用甲醇稀释至刻度,保存于4 ℃冰箱中(不超过3个月)。
试剂处理:DEAEMA用CaH2干燥48 h,减压蒸馏后储存于-20 ℃冰柜中;PEGMA过中性氧化铝层析柱,除去阻聚剂后密封待用;甲苯、三乙胺、二氯甲烷等均经干燥处理,待用。
1.3 亲水性聚合物刷磁性微球的制备
1.3.1 磁性微球的制备
采用共沉淀法合成磁性微球。称取2.00 g FeCl254H2O和5.20 g FeCl356H2O,加入100.0 mL去离子水,机械搅拌使其充分溶解,在氮气条件下加热至80 ℃,将40.0 mL 0.20 mol/L氢氧化钠溶液逐滴滴加到混合物中,溶液由棕黄色变为黑色后将2.0 mL油酸滴加到溶液中,于80 ℃恒温反应1 h,用乙醇和蒸馏水分别洗涤产物3次,除去未反应的化合物,于60 ℃下干燥12 h,得到Fe3O4磁性微球(Fe3O4-MMs)。
1.3.2 硅胶包覆磁性微球的制备
利用溶胶-凝胶法合成硅胶包覆的磁性微球。取1.00 g Fe3O4-MMs,加入80.0 mL乙醇、25.0 mL蒸馏水和2.5 mL氨水,超声分散5 min。在机械搅拌条件下将1.0 mL TEOS与20.0 mL乙醇的混合溶液逐滴加入上述体系中,超声分散5 min。于35 ℃机械搅拌12 h,收集产物。用乙醇和蒸馏水分别洗涤3次,再用1.00 mol/L的盐酸浸泡12 h,用蒸馏水洗涤至中性,于60 ℃干燥12 h。然后于150 ℃真空干燥箱中活化3 h,除去微球表面的缔合水,得到硅胶包覆的磁性微球MMs@SiO2。将制备好的MMs@SiO2保存在真空干燥器中。
1.3.3 氨基功能化磁性微球的制备
通过甲苯回流法制备氨基功能化磁性微球。称取1.00 g MMs@SiO2,加入100.0 mL甲苯,超声分散,在氮气保护下,加入10.0 mL APTES,超声5 min,于110 ℃机械搅拌条件下回流反应24 h。产物通过磁铁分离,用乙醇和蒸馏水分别洗涤3次,于40 ℃真空干燥箱中干燥12 h,最后得到氨基功能化磁性微球MMs@SiO2-NH2。
1.3.4 磁性微球表面接枝引发剂
在干燥的250 mL圆底烧瓶中加入70.0 mL二氯甲烷,将1.00 g干燥的MMs@SiO2-NH2磁性微球超声分散于二氯甲烷中。加入2.0 mL干燥的三乙胺,并用冰水浴将体系冷却至0 ℃。将2.0 mL 2-溴异丁酰溴溶于30.0 mL二氯甲烷中。在冰水浴和机械搅拌条件下,将上述混合溶液逐滴加入圆底烧瓶中,混合体系在冰水浴条件下继续机械搅拌反应2 h,然后撤去冰浴,在室温条件下机械搅拌12 h。反应结束后,得到的产物依次用无水乙醇和蒸馏水洗涤数次,于40 ℃真空干燥箱中干燥12 h,制备得到表面接枝引发剂的磁性微球MMs-initiator,将其保存在真空干燥器中。
1.3.5 聚合物刷磁性微球的制备
参照文献[19]中亲水嵌段胶束的合成方法,在磁性微球表面接枝了聚碱类聚合物和聚乙二醇刷。在100 mL干燥圆底烧瓶中加入1.00 g MMs-initiator和0.09 g CuBr2,密封反应瓶,通N2排尽空气。依次将90.0 mL无水甲苯、19.5 mL DEAEMA单体和835 μL PMDETA配体加入反应瓶中,搅拌10 min,使催化剂配合物Cu-PMDETA形成。随后将1.3 mL还原剂Sn(Oct)2溶于10.0 mL甲苯中,混合均匀后加入反应瓶中,机械搅拌5 min,然后将反应瓶转入70 ℃油浴中,机械搅拌反应7 h。为了对比研究的需要,反应7 h后,取出一部分冷却至室温,依次用无水乙醇和蒸馏水洗涤3次,于40 ℃真空干燥箱中干燥12 h,获得的产物为接枝了聚碱类聚合物的磁性微球DMMs。在余下的反应体系中,加入8.3 mL PEGMA,连续聚合8 h后,冷却至室温,得到内层为聚碱类聚合物、外层为亲水性聚合物刷的磁性微球HMMs。将HMMs依次用无水乙醇和蒸馏水洗涤3次,于40 ℃真空干燥箱中干燥12 h后,保存在真空干燥器中。
1.4 静态吸附试验
将20 mg DMMs和HMMs分别置于25 mL具塞锥形瓶中,依次用3.0 mL甲醇和3.0 mL高纯水进行洗涤活化,然后在10.0 mL含有不同质量浓度(0.01~0.20 g/L)TC的B-R缓冲溶液Ⅰ中进行吸附试验,室温下摇床振荡萃取60 min。萃取结束后,经磁分离,用紫外分光光度计在365 nm处检测萃取后各溶液中TC的质量浓度。
分别称取10 mg LZM和PRM,用10 mL B-R缓冲液Ⅱ溶解,按照上述步骤萃取后,移取上清液0.1 mL,加入5 mL考马斯亮蓝试剂,振荡混匀,在595 nm处检测各蛋白质的质量浓度。
通过公式Q=V(Co-Ce)/m计算平衡吸附容量Q(mg/g),其中,V(mL)是溶液的体积;Co(g/L)和Ce(g/L)分别是溶液中药物的初始质量浓度和萃取平衡后的质量浓度;m(mg)为聚合物刷磁性微球的质量。
1.5 样品前处理
称取5.00 g蜂蜜样品,置于锥形瓶中,加入25.0 mL Na2EDTA-Mcllvaine缓冲溶液,样品完全溶解后,旋蒸至1.0 mL左右。然后用B-R缓冲溶液Ⅰ定容至10.0 mL。
取20 mg HMMs置于锥形瓶中,依次用3.0 mL甲醇和3.0 mL水进行活化,然后加入10.0 mL上述提取的样品溶液,于室温下振荡萃取30 min,磁铁分离后倾去上清液。用3.0 mL二氯甲烷淋洗HMMs,然后用3.0 mL B-R缓冲溶液Ⅱ进行洗脱,磁铁分离后,将洗脱液氮吹至近干,用1.0 mL流动相重新溶解,经0.22 μm的有机滤膜过滤后,待HPLC分析。
1.6 高效液相色谱条件
色谱柱:Venusil XBP C18柱(250 mm×4.6 mm, 5 μm,天津博纳艾杰尔科技有限公司);柱温:25 ℃;流动相A: 0.01 mol/L草酸溶液;流动相B:乙腈-甲醇(1∶1, v/v);梯度洗脱(具体条件见表1);流速:1.00 mL/min;检测波长:365 nm;进样量:20 μL。流动相和样品均经0.22 μm有机滤膜过滤后使用。

表 1 梯度洗脱条件Table 1 Gradient elution conditions
A: 0.01 mol/L oxalic acid solution; B: acetonitrile-methanol (1∶1, v/v).
2 结果与讨论
2.1 聚合物刷磁性微球的表征
图1a~1c分别为Fe3O4-MMs、DMMs和HMMs的透射电镜图。由图1a可知,制备的Fe3O4-MMs分布较为均匀,平均粒径约为180 nm;由图1b可知,在Fe3O4-MMs表面接枝了DEAEMA,其聚合物层的平均厚度为10 nm;由图1c可知,继续接枝PEGMA后,HMMs聚合物层的厚度达到40 nm。结果表明,HMMs被成功合成。

图 1 (a)Fe3O4磁性微球、(b)聚碱类聚合物磁性微球和(c)亲水性聚合物刷磁性微球的透射电镜图Fig. 1 Transmission electron microscopy images of (a) Fe3O4 magnetic microspheres (Fe3O4-MMs), (b) polybasic polymers magnetic microspheres (DMMs) and (c) hydrophilic polymer brushes magnetic microspheres (HMMs)
Fe3O4-MMs、MMs@SiO2、MMs@SiO2-NH2、MMs-initiator、DMMs和HMMs的傅里叶变换红外光谱图见图2,通过傅里叶变换红外光谱图可以进一步验证不同磁性微球表面基团的接枝情况。从图2a可以看出,在587 cm-1左右有吸收峰,这是Fe3O4-MMs中 Fe=O 的伸缩振动峰,证明成功合成了Fe3O4-MMs;在1 094 cm-1和467 cm-1处分别为 Si-O-Si 的非对称伸缩振动峰和对称伸缩振动峰,并且在587 cm-1处Fe3O4的特征峰变小,说明硅胶成功包覆在Fe3O4磁性微球表面(见图2b);与图2b相比,图2c中多了1 632 cm-1处 N-H 的伸缩振动峰和2 963 cm-1处 -CH2- 的伸缩振动峰,表明成功合成了MMs@SiO2-NH2;图2d中,-Br的特征峰在700~500 cm-1附近,本实验中和Fe3O4的特征峰发生重合,故无法观察到,但与图2c相比,多了1 653 cm-1处酰胺基团中 C=O 的伸缩振动峰,证明在磁性微球表面成功接枝了2-溴异丁酰溴引发剂;图2e中多了1 720 cm-1处的 C=O 伸缩振动峰、1 380 cm-1处 -CH3的特征峰和1 459 cm-1处 -CH2- 的变形振动峰,这些均表明成功制备了DMMs;从图2f可以看出,3 443 cm-1处 O-H 的伸缩振动峰、1 720 cm-1处 C=O 伸缩振动峰、1 380 cm-1处 -CH3的特征峰和1 459 cm-1处 -CH2- 的变形振动峰都明显增大,从而证明成功合成了HMMs。
表征了不同磁性微球的热稳定性情况。Fe3O4-MMs、MMs@SiO2、MMs@SiO2-NH2、MMs-initiator、DMMs和HMMs的热重分析图见图3。从图3可以看出,Fe3O4-MMs和MMs@SiO2的质量损失分别为4.64%和6.15%,表明了其稳定性良好。MMs@SiO2-NH2有7.39%的质量损失,而MMs-initiator的质量损失达到11.36%,这就表明约有4.00%的2-溴异丁酰溴通过酰胺反应接枝到磁性微球表面。从DMMs的热重分析曲线可以看出,DMMs的质量损失较大,可达到18.35%,可能是由于高分子聚合物聚甲基丙烯酸二乙氨基乙酯(PDEAEMA)的分解所致。接枝了PEGMA聚合物刷的HMMs在0~800 ℃之间质量损失增加到32.73%。通过图3的热重曲线可知,样品的质量损失逐步增加,说明每个步骤都反应成功。
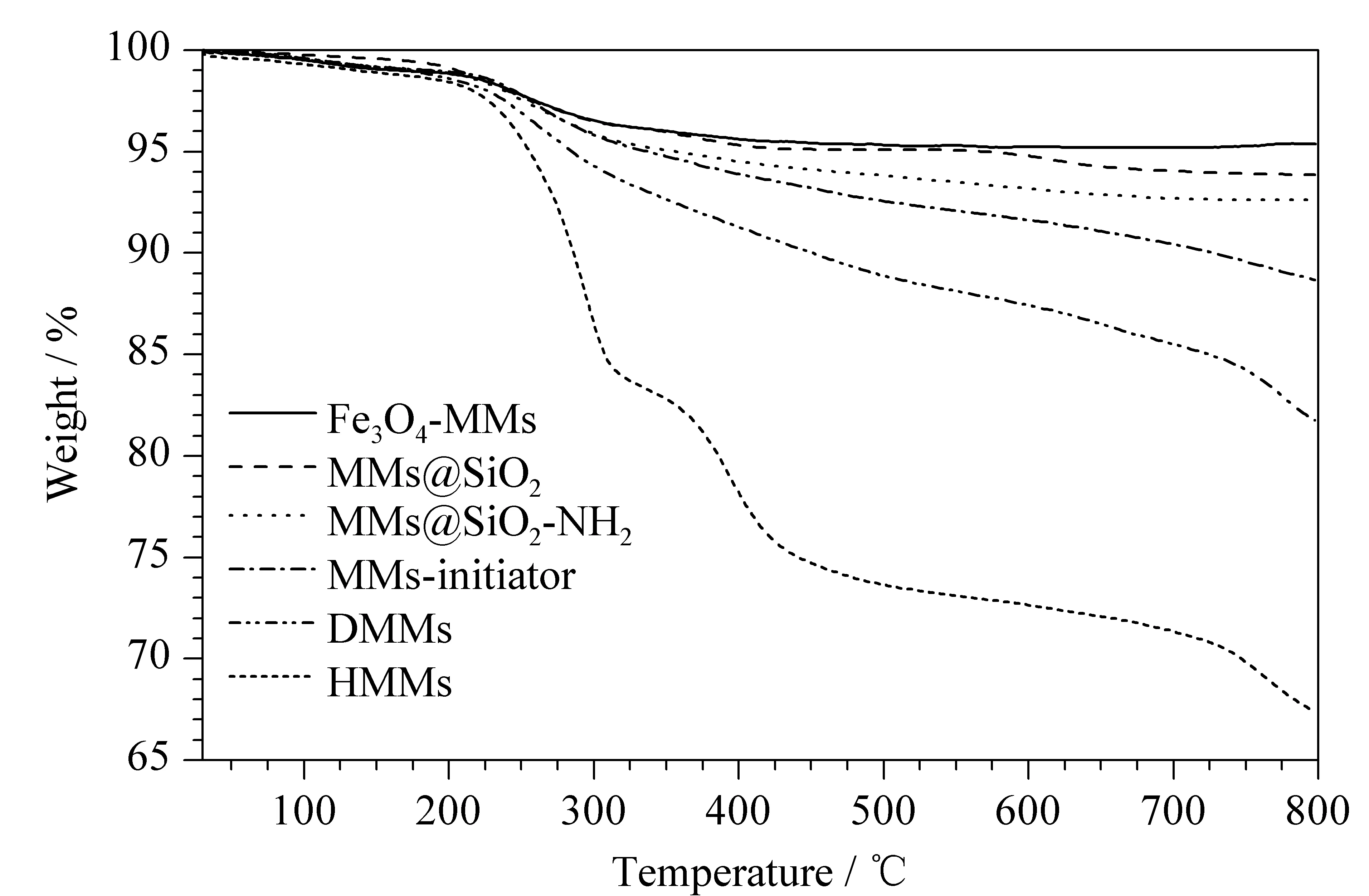
图 3 不同磁性微球的热重分析曲线Fig. 3 Thermogravimetric analysis curves of the different magnetic microspheres
2.2 静态吸附考察
2.2.1 对TC的吸附性能考察

图 4 DMMs和HMMs对(a)盐酸四环素和(b)蛋白质的吸附性能(n=3)Fig. 4 Adsorption performances of DMMs andHMMs for (a) tetracycline hydrochloride (TC) and (b) proteins (n=3) PRM: protamine; LZM: lysozyme.
图4a为25 ℃时,DMMs和HMMs对不同质量浓度(0.01~0.20 g/L)TC溶液的吸附性能曲线。由图4a可知,当TC的质量浓度小于0.05 g/L时,HMMs对TC的吸附量随着TC质量浓度的增大而增大;当TC的质量浓度大于0.05 g/L时,HMMs对TC的吸附量基本保持不变,说明达到了吸附平衡,平衡时吸附量为10.51 mg/g。当TC的质量浓度大于0.05 g/L时,DMMs对TC的吸附量仍处于增长趋势,直至TC的质量浓度为0.11 g/L后才达到吸附平衡。在达到吸附平衡时,DMMs对TC的吸附量大于HMMs的吸附量,这是由于DMMs表面与TC存在着较强的静电作用和疏水作用,接枝亲水性聚合物刷后,降低了聚碱类聚合物和TC的作用位点。但HMMs的吸附量远大于实际样品中TCs的含量。
2.2.2 对蛋白质的吸附性能考察
采用1.4节静态吸附的试验步骤,研究了DMMs和HMMs对蛋白质的吸附性能。实验选用的蛋白质为PRM和LZM,并利用考马斯G-250法检测萃取前、后蛋白质的含量。结果如图4b所示:DMMs对蛋白质有很强的吸附,接枝了亲水性聚合物刷后,吸附剂HMMs对蛋白质的吸附量明显降低,表明HMMs对蛋白质表现出良好的排阻性能。这主要归因于亲水长链的弹簧效应,当蛋白质靠近并压缩亲水性长链时,长链会像弹簧一样对蛋白质产生排斥作用,使得蛋白质不能吸附在材料表面。随着亲水性长链接枝密度增加以及链段长度的增大,蛋白质的吸附量会随之减少[20]。
综上所述,HMMs具有较好的样品净化能力和良好的吸附性能,因此本研究选择HMMs为吸附剂进行磁分散固相萃取,并用于实际样品的分离分析研究。

图 5 (a)上样溶液的pH值、(b)萃取时间和(c)淋洗溶剂、洗脱剂对TC(0.05 g/L)萃取效率的影响(n=3)Fig. 5 Effects of (a) the pH value of loading solvents(b) extraction time and (c) washing solvents on the extraction efficiencies of TC (0.05 g/L, n=3) The washing solvents and elution solvents in Fig. 5c: 1. methanol-acetic acid (4∶1, v/v); 2. methylbenzene; 3. methanol-methylbenzene (1∶19, v/v); 4. dichloromethane; 5. ethyl acetate; 6. alcohol; 7. tetrahydrofuran; 8. Britton-Robinson buffer solution Ⅱ(0.022 mol/L of phosphoric acid-acetic acid-boric acid and 0.087 mol/L of sodium hydroxide, pH 10.0).
2.3 磁分散固相萃取条件的优化
以HMMs为吸附剂,对磁分散固相萃取的条件(上样溶液的pH、萃取时间、淋洗液和洗脱剂)进行了详细优化(见图5)。
2.3.1 上样溶液的pH对萃取效率的影响
在25 ℃条件下,以不同pH的B-R缓冲溶液为上样溶液(TC的质量浓度为0.05 g/L),考察pH值对TC萃取效率的影响。由图5a可知,以HMMs为吸附剂时,吸附量随着pH值的增加呈先增大后减小的趋势,并且在pH=6.00时吸附量达到最大。因此实验选择pH=6.00的B-R缓冲溶液Ⅰ为上样溶液。
2.3.2 萃取时间对萃取效率的影响
考察了在25 ℃、pH=6.00和TC质量浓度为0.05 g/L时萃取时间对萃取效率的影响。如图5b所示,萃取时间为0~30 min时,HMMs对TC的吸附量随萃取时间的增加明显增大,在30 min时吸附量达到最大;继续增加萃取时间,吸附量没有明显变化,即吸附已达到饱和。因此实验选择30 min为实际样品的萃取时间。
2.3.3 淋洗液和洗脱剂对萃取效率的影响
淋洗液的目的是洗去杂质,保留目标物质;洗脱剂的目的是将目标物质洗脱下来。因此,为了更好的避免样品中复杂基质的干扰同时将目标物质更好地洗脱下来,选择8种不同极性的溶剂对HMMs上的TC进行解吸附。由图5c可以看出,当选择二氯甲烷溶液为淋洗溶剂时,TC的回收率最低,因此,选择二氯甲烷溶液作为淋洗液;当B-R缓冲溶液Ⅱ为淋洗溶剂时,TC的回收率最高,因此实验选择B-R缓冲溶液Ⅱ为洗脱剂。
2.4 吸附剂的重复性考察
为了研究HMMs的可重复性,称取20 mg活化后的HMMs,加入10.0 mL质量浓度为0.05 g/L的TC溶液,按1.5节步骤进行磁分散固相萃取。将吸附剂HMMs循环使用7次,每次3组平行试验。实验结果如图6所示:第5次循环吸附的平均吸附容量为9.61 mg/g,与第一次吸附容量相比,仍能保持在原吸附容量的(93.00±1.00)%;第5~7次间的吸附量相差较少,并趋于稳定。通过循环试验证明了HMMs吸附剂具有较好的吸附与解吸附性质,可重复使用。
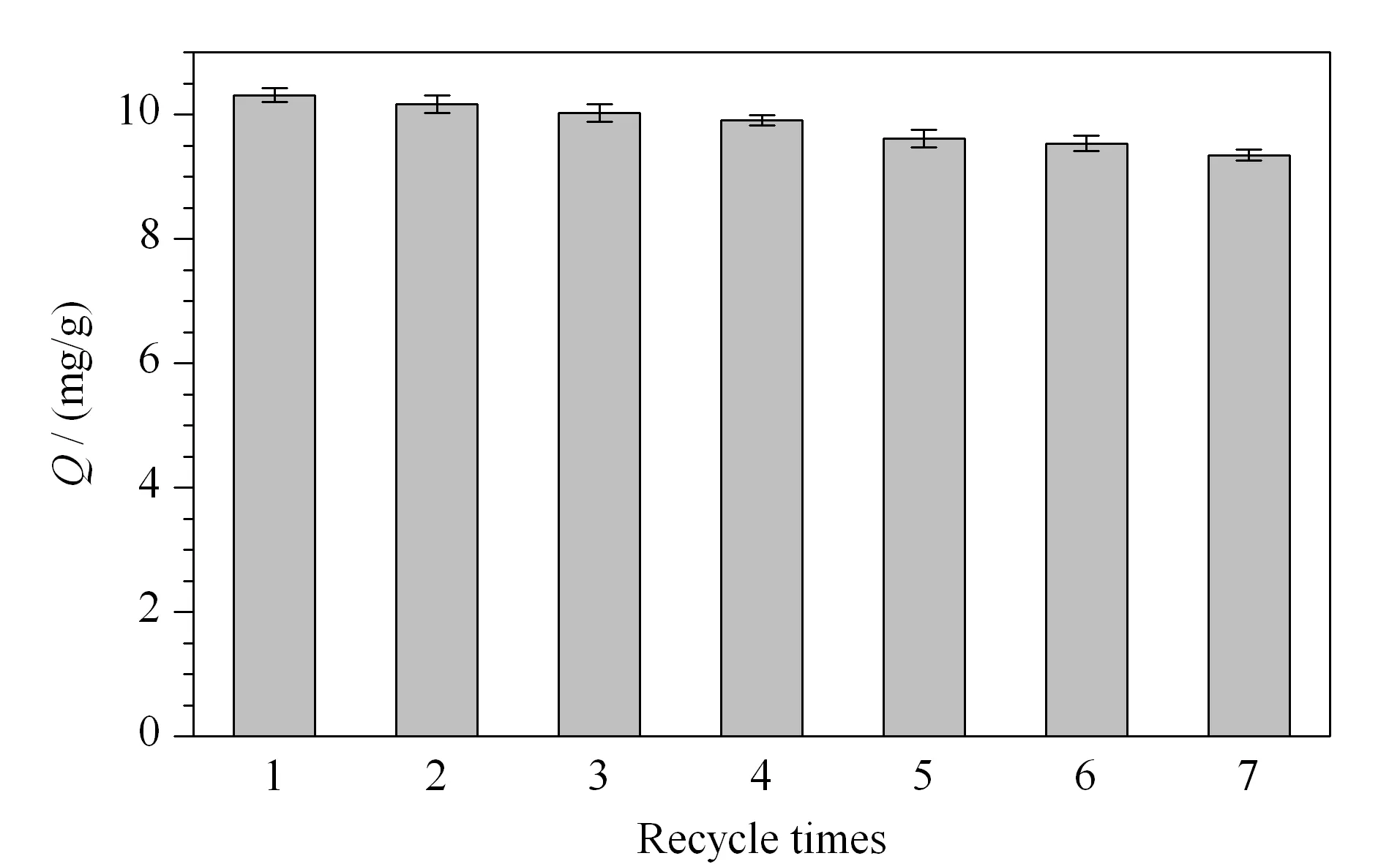
图 6 HMMs的可重复性(n=3)Fig. 6 Reusability of the HMMs (n=3)

图 7 (a)混合标准溶液(0.05 mg/L)、(b)空白蜂蜜样品和(c)加标(100 μg/kg)蜂蜜样品中四环素类抗生素的色谱图 Fig. 7 Chromatograms of the tetracycline antibiotics(TCs) in (a) a mixed standard solution (0.05 mg/L), (b) a black honey sample and (c) a spiked honey sample (100 μg/kg)
2.5 富集与净化效果的考察
为了研究HMMs的富集与净化效果,分别对质量浓度为0.05 mg/L的TCs(TC、CTC和DC)混合标准溶液(见图7a)、空白蜂蜜样品(见图7b)和加标(100 μg/kg)蜂蜜样品(见图7c)进行磁分散固相萃取,洗脱液经氮吹至近干后用1.0 mL的流动相溶解,最后经HPLC检测。从图7a可以看出,磁分散固相萃取前,TCs的色谱峰信号较弱,难以定量;经磁分散固相萃取后,目标物的色谱峰信号增强;由图7b和7c可知,经磁分散固相萃取处理后,色谱图不但减少了杂质峰的干扰,基线漂移降低,而且富集后目标物的色谱峰信号明显增强。由此可见,合成的HMMs对蜂蜜样品中的TCs有较好的净化和富集效果。
2.6 线性方程和检出限
配制系列质量浓度为0.02、0.025、0.05、0.10、0.20、0.50和1.00 mg/L的TCs混合标准溶液,在1.6节所述的色谱条件下进行检测,得到3种物质的线性方程(见表2)。结果显示,TC、CTC和DC的峰面积(A)与质量浓度(C, mg/L)在0.02~1.00 mg/L范围内均表现出良好的线性关系,相关系数(R2)为0.999 1~0.999 9。TCs的检出限(LOD,S/N=3)和定量限(LOQ,S/N=10)分别为1.92~2.56 μg/kg和6.40~8.53 μg/kg。
2.7 富集因子
富集因子(enrichment factor, EF)可通过方程式EF=Co/Ci计算得到,其中,Co(g/L)和Ci(g/L)分别是样品溶液中分析物的初始质量浓度和经MDSPE后的质量浓度。经计算,TC、CTC和DC的平均富集因子分别为7.59、3.73和4.03(见表2)。

表 2 3种TCs的线性方程、线性范围、相关系数、检出限、定量限和富集因子Table 2 Linear equations, linear ranges, correlation coefficients (R2), limits of detection (LODs), limits of quantification (LOQs) and enrichment factors (EFs) of the three TCs
A: peak area;C: mass concentration, mg/L.
2.8 实际样品分析
以HMMs为磁性微球,采用MDSPE-HPLC测定蜂蜜样品中的TCs,并进行了蜂蜜样品的加标回收试验。如表3所示,在0.05、0.10和0.20 mg/kg的加标水平下,TC、CTC和DC的回收率为85.8%~94.5%,相对标准偏差为1.6%~4.4%。结果表明,制备的HMMs能够用于食品和生物等复杂基质样品中TCs的净化和富集。

表3 加标蜂蜜样品中3种TCs经磁分散固相萃取后的回收率和相对标准偏差(n=3)Table3 Recoveriesandrelativestandarddeviations(RSDs)ofthethreeTCsspikedinthehoneysamplesaftermagneticdispersionsolidphaseextraction(MDSPE)(n=3) AnalyteSpikedlevel/(mg/kg)Detected/(mg/kg)Recovery/%RSD/%TC0.050.047294.51.70.100.093993.91.90.200.018190.52.5CTC0.050.046993.71.60.100.088288.22.80.200.181290.63.3DC0.050.045791.34.40.100.092192.13.20.200.171685.82.7
3 结论
本文采用电子转移活化再生催化剂原子转移自由基聚合法,经过连续聚合反应制备了亲水性聚合物刷磁性微球。制备的亲水性聚合物刷磁性微球形状规则,大小较为均一,并且具有良好的蛋白质排阻能力。将制备的亲水性聚合物刷磁性微球作为磁分散固相萃取吸附剂用于分离富集蜂蜜中四环素类抗生素,得到了良好的富集净化效果。该亲水性聚合物刷磁性微球在分离富集和净化生物样品、食品样品和环境样品的应用中具有较大潜力。
[1] Code of Federal Regulations (CFR), Title 21, Part 556. Tolerances for Residues of New Animal Drugs in Food. (2013-04-01) [2017-11-18]. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=820& showFR=1
[2] 2377/90/EEC
[3] Ministry of Agriculture. No. 235 Bulletin of the Ministry of Agriculture of the People’s Republic of China. (2002-12-24) [2017-11-18]. http://yz.hz-agri.gov.cn/uploadFiles/2005-10/1130221564406.doc
农业部. 中华人民共和国农业部公告第235号. (2002-12-24) [2017-11-18]. http://yz.hz-agri.gov.cn/uploadFiles/2005-10/1130221564406.doc
[4] GB/T 22990-2008
[5] Phiroonsoontorn N, Sansuk S, Santaladchaiyakit Y, et al. J Chromatogr A, 2017, 1519: 38
[6] Lv Y K, Wang L M, Yang L, et al. J Chromatogr A, 2012, 1227(5): 48
[7] GB/T 23409-2009
[8] Xu H, Mi H Y, Guan M M, et al. Food Chem, 2017, 232: 198
[9] Guo M, Yu F, Jia K L, et al. Chinese Journal of Chromatography, 2016, 34(4): 407
国明, 于峰, 贾科玲, 等. 色谱, 2016, 34(4): 407
[10] Rios A, Zougagh M, Bouri M. Anal Methods, 2013, 5(18): 4558
[11] Li X S, Zhu G T, Luo Y B, et al. TrAC-Trends Anal Chem, 2013, 45(4): 233
[12] Lv Y K, Zhao C X, Li P, et al. J Sep Sci, 2013, 36(16): 2656
[13] Lv Y K, He Y D, Xiong X, et al. New J Chem, 2015, 39(3): 1792
[14] Lv Y K, Zhang J, Li M Z, et al. Anal Methods, 2016, 8(19): 3982
[15] Lv Y K, Xiong X, Zhao F F, et al. Aust J Chem, 2017, 70(3): 237
[16] Cao X T, Horak D, An Z S, et al. J Polym Sci Pol Chem, 2016, 54(8): 1036
[17] Yang Y, Liu X, Ye G, et al. ACS Appl Mater Inter, 2017, 9(15): 13637[18] Xie L, Lan F, Li W, et al. Colloids Surf B Biointerfaces, 2014, 123: 413
[19] Yang Y Q, Zhao B, Li Z D, et al. Acta Biomater, 2013, 9(8): 7679
[20] Inoue Y, Nakanishi T, Ishihara K. React Funct Polym, 2011, 71(3): 350
猜你喜欢
化学工业与工程(2022年1期)2022-03-29 01:14:32
合成树脂及塑料(2020年6期)2020-12-29 07:02:02
化学与粘合(2020年6期)2020-03-08 09:06:30
石油沥青(2019年4期)2019-09-02 01:41:54
科技视界(2017年25期)2017-12-11 20:30:32
中国塑料(2016年3期)2016-06-15 20:30:03
中国学术期刊文摘(2016年2期)2016-02-13 16:01:41
中国塑料(2015年1期)2015-10-14 00:58:41
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:50
河南科技(2014年15期)2014-02-27 14:12:29
